
Immune Assembly (Part 1)
Lymph-Node Priming as the Foundation of Durable Immunotherapy

There is a phrase that appears again and again in conversations about immunotherapy failure. It’s usually spoken gently, often by smart people, and almost always with good intentions: “Lymph-node priming is interesting, but it’s still a bit speculative.” That sentence used to be defensible.
It no longer is. What follows is not a claim of novelty, nor an argument from enthusiasm. It is a record of convergence—across mechanistic immunology, spatial biology, translational oncology, and late-stage clinical pattern recognition—that has quietly but decisively closed the debate.
Lymph-node (LN) priming is not a hypothesis in search of evidence. It is a contested control layer in anti-tumor immunity, and its integrity—or collapse—now explains more about checkpoint success and failure than PD-L1, TMB, or target engagement alone—each of which still modifies probability, but cannot compensate for collapsed immune assembly.
The Persistent Confusion: Target Failure vs. Infrastructure Failure
Checkpoint inhibitors were built on an elegant and deeply intuitive premise: remove the inhibitory signal, and the immune system—already primed, already competent—will do the rest. When this works, the story feels almost tautological. Tumors regress. Memory forms. The immune system appears, retrospectively, as though it had simply been waiting for permission.
But when it doesn’t work—and for most patients, it doesn’t—the field has spent more than a decade circling the wreckage, arguing about why. Is the tumor “cold”? Is antigen missing or poorly presented? Is PD-L1 mismeasured, transient, or spatially misleading? Is the patient somehow “non-inflamed,” as if inflammation were a personal attribute rather than a biological state?
Each of these explanations carries a grain of truth. And yet, taken together, they share a common limitation: they are all endpoint explanations. They interrogate what is visible at the tumor site after therapy has failed. They diagnose absence where effect was expected. They circle the tumor, again and again, asking why the immune response did not appear there. What they rarely ask is the more uncomfortable upstream question: was the immune response ever structurally capable of forming in the first place? Recent reviews have framed tumor-draining lymph nodes as both facilitators and inhibitors of immunity, a duality that likely contributed to earlier conceptual ambiguity rather than true biological uncertainty.
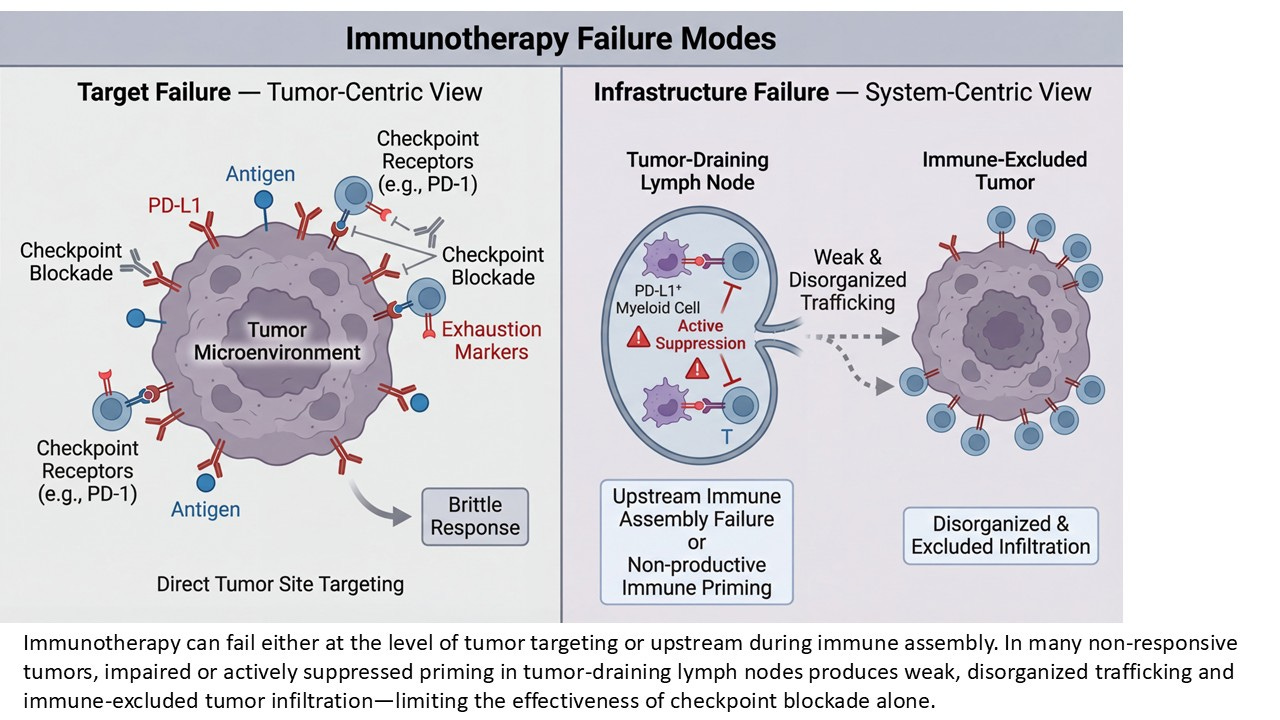
This is the question lymph-node priming forces into view. Rather than treating the tumor as the origin of immune failure, LN priming reframes failure as a problem of infrastructure, not intent. It asks whether the immune system had the architectural prerequisites required to generate a durable, amplifiable response—long before PD-1 blockade ever entered the picture.
This shift matters. It moves the axis of inquiry away from static tumor snapshots and toward immune choreography: the sequence of spatial, cellular, and temporal events that must occur upstream for checkpoint inhibition to have anything meaningful to amplify. In this framing, checkpoint inhibitors are no longer the initiators of immunity. They are late-stage accelerants. And like any accelerant, they are powerless if the underlying structure was never built. Failure, then, is not always a question of missing targets or insufficient inflammation at the endpoint. Sometimes it is the quieter, more consequential absence of a system that was never assembled.
What LN Priming Actually Means (and What It Doesn’t)
Lymph-node priming has suffered less from lack of evidence than from excess metaphor. Over time, it has been used to gesture at everything from immune “activation” to antigen exposure to vague notions of immune readiness. In that diffusion, its meaning has blurred—and with it, its explanatory power. So it is worth being precise.
LN priming is not a synonym for immune stimulation. It is not a measure of cytokine release.
It is not a proxy for tumor inflammation, nor is it interchangeable with T-cell presence in the tumor microenvironment. LN priming refers to a specific sequence of spatially anchored events that must occur upstream of the tumor for adaptive immunity to become both effective and durable.
At its core, it describes whether tumor-derived antigens are successfully trafficked to tumor-draining lymph nodes, whether dendritic cells are appropriately licensed and positioned, and whether naïve and progenitor T cells are given the conditions necessary to differentiate without prematurely exhausting. It encompasses not just activation, but instruction—the difference between generating effector cells that burn out quickly and preserving stem-like populations capable of sustaining response over time.
This distinction matters because the lymph node is not simply a relay station. It is a decision-making environment. Signals received there—co-stimulatory cues, cytokine gradients, the balance of activating versus suppressive cells—shape the fate of the immune response long before a T cell ever encounters a tumor cell again.
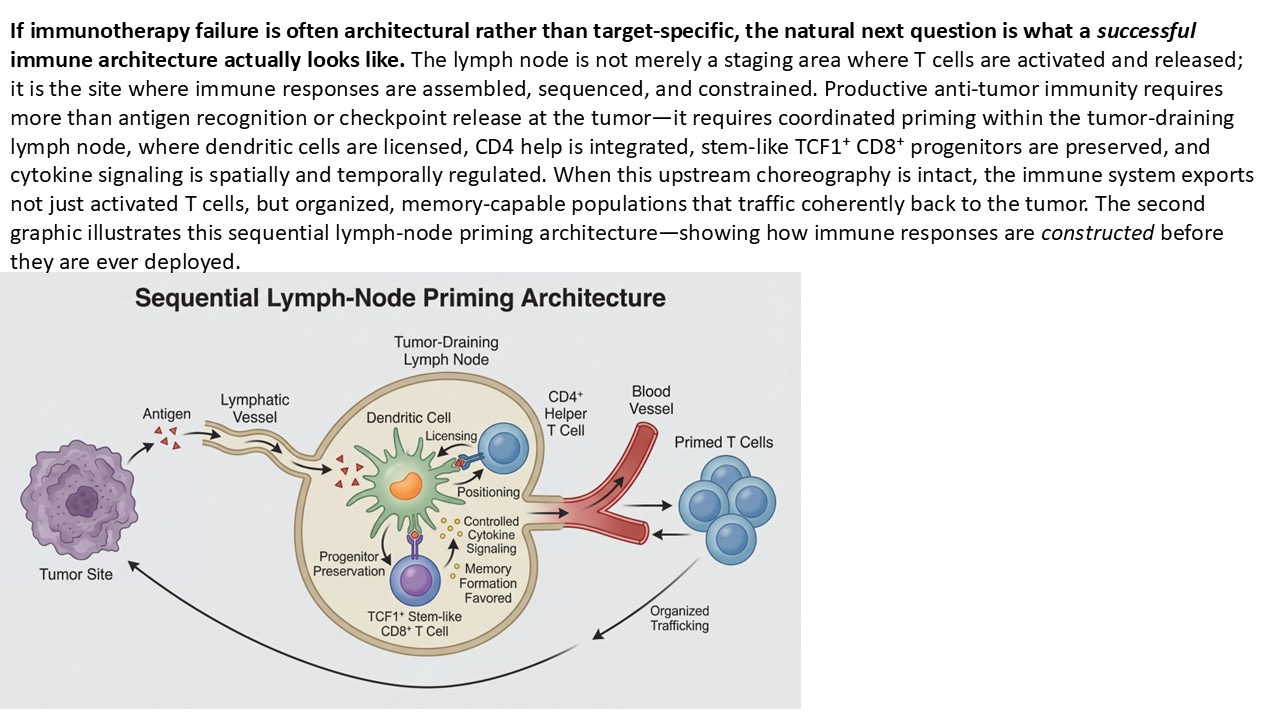
When LN priming functions properly, it produces a population of T cells that can expand, traffic, persist, and respond to amplification. When it fails, the immune system may still generate signals that look like activity—transient inflammation, scattered infiltration, even partial responses—but these signals lack structural support. They cannot be reliably amplified, and they rarely endure. Recent multi-omics analyses further reinforce this view, positioning tumor-draining lymph nodes as integrative hubs where antigen presentation, progenitor fate, and suppressive programming can be jointly interrogated rather than inferred.
Equally important is what LN priming is not. It is not a claim that the tumor microenvironment is irrelevant. Tumors still erect barriers, exclude immune cells, and apply local suppression. But those barriers are often secondary. They determine how a response is expressed, not whether one can exist at all. Nor does LN priming imply that checkpoint blockade is unnecessary or obsolete. Rather, it clarifies checkpoint inhibitors’ true position in the immune sequence: they act on a response that has already been built. If that response was malformed or incomplete at the priming stage, removing inhibitory signals downstream cannot compensate for what was never assembled.
In this sense, LN priming is not an alternative theory of immunotherapy. It is the structural layer beneath all of them—a prerequisite that explains why the same checkpoint inhibitor can produce durable remission in one patient and no response in another, even when tumors appear superficially similar. Understanding LN priming, then, is not about adding complexity for its own sake. It is about restoring causal order to a field that has too often mistaken late-stage amplification for immune genesis.
The Tumor-Side Clue We Missed for Years: eTIL Patterning
For years, tumor-infiltrating lymphocytes were treated as a blunt instrument. More was assumed to be better. Less was assumed to be worse. And somewhere along the way, “TIL-high” and “TIL-low” hardened into crude labels that promised clarity but delivered very little. The problem was not that TILs were irrelevant. It was that we were asking the wrong question of them.
Counting lymphocytes inside a tumor answers a superficial query: is the immune system present?
What it does not answer is the more consequential one: how did it get here, and in what state?
Recent work—particularly from the last several years—has finally reframed the issue. Across multiple tumor types, response to checkpoint blockade correlates far more consistently with the spatial organization and functional patterning of tumor-infiltrating lymphocytes than with raw abundance alone. Where T cells reside, how deeply they penetrate, whether they engage the tumor–stroma interface, and whether they form coherent immune niches all matter more than the absolute number counted in a field.
Immune-inflamed tumors—those with parenchymal penetration and structured interface engagement—behave fundamentally differently from immune-excluded tumors, even when both contain lymphocytes. Excluded tumors often carry the visual suggestion of immune presence without the functional reality of immune control. They represent responses that arrived fragmented, misdirected, or already exhausted. This distinction is not cosmetic. It is causal.
What eTIL patterning reveals is not simply the tumor’s permissiveness, but the history of the immune response upstream. Tumor infiltration is not an autonomous event. T cells do not simply appear in tissue because checkpoints are removed. They arrive as the downstream consequence of successful priming, expansion, and trafficking—processes that are orchestrated in the lymph node.
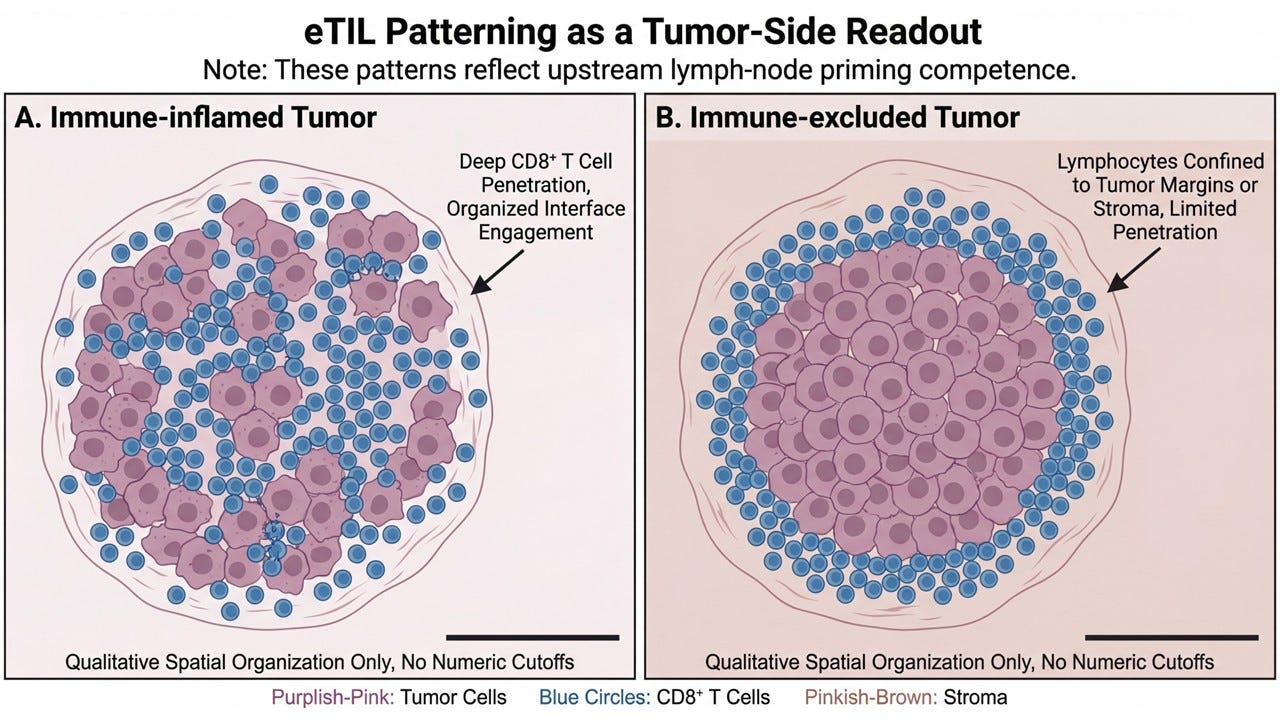
Seen this way, eTIL patterning becomes something more than a prognostic marker. It becomes a tumor-side readout of lymph-node competence. When LN priming is intact, the immune response tends to organize. Progenitor populations are preserved. Effector cells traffic with purpose. Tumor penetration follows recognizable spatial rules. When LN priming is impaired, infiltration—if it occurs at all—appears chaotic, peripheral, or stalled at the margins.
This explains a long-standing paradox in immunotherapy: why some tumors that technically contain lymphocytes behave as though they are immunologically inert. The issue is not presence. It is provenance. The tumor, in other words, is not the origin of the failure. It is the mirror. And for a long time, we mistook the reflection for the cause.
Clinical Pattern Recognition: TCF1⁺ Persistence as a Living Signal of LN Priming
One of the quiet strengths of the LN priming framework is that it does not require new clinical behavior to be true — it only requires us to recognize patterns that have already been observed but misinterpreted.
ABILITY-1 offers one of the clearest examples. Across treated patients, MDNA11 exposure was associated with the persistence of TCF1⁺ CD8⁺ populations over time, rather than their rapid differentiation into terminal effector states. This is not a trivial observation. TCF1⁺ cells are not markers of generic immune activation. They are markers of instructional quality — of whether CD8⁺ T cells were primed under conditions that preserved renewal capacity rather than forced early exhaustion.
What makes this clinically meaningful is not that TCF1⁺ cells were detectable at baseline — many patients harbor such cells somewhere. What matters is that they persisted across dosing cycles, implying that upstream conditions continued to support renewal rather than collapse. That persistence is difficult to explain if immune activity is being driven primarily by intratumoral reinvigoration. Terminally exhausted cells, once pushed, do not regenerate themselves. They burn.
Persistence, by contrast, implies a functioning upstream source. Within the LN priming framework, this pattern reads clearly: the tumor-draining lymph node remained permissive to productive instruction. Antigen presentation, dendritic licensing, cytokine signaling, and progenitor fate decisions remained aligned long enough to support ongoing export. Effector waves could be generated because the substrate was not exhausted.
This also explains a second ABILITY-1 observation that has been easy to overlook: kinetics varied without collapsing durability expectations. Some patients showed delayed tumor responses without biomarker chaos. In an architecture-first model, that delay is not noise — it is construction time. Progenitor pools do not rebuild instantaneously. Trafficking does not normalize in a single burst. But once renewal is re-established, responses stabilize.
Seen this way, TCF1⁺ persistence is not a downstream correlate of response. It is a living signal that LN priming has been restored or preserved, even when radiographic confirmation lags. ABILITY-1 did not “discover” LN priming — it inadvertently validated it.
That distinction matters, because it shifts TCF1⁺ biology from exploratory curiosity to mechanistic confirmation of upstream immune assembly at work.
The 2024–2025 Shift: When LN Suppression Becomes Mechanistic, Not Speculative
For years, lymph-node priming carried an implicit vulnerability. Even as evidence accumulated, critics could still dismiss it with a familiar refrain: interesting in theory, but difficult to pin down mechanistically. The lymph node, after all, is upstream, dynamic, and rarely biopsied. Its failures were inferred, not observed. That gap has now narrowed. Notably, early-phase clinical efforts are now explicitly targeting lymph-node–localized immune programming, underscoring a shift from downstream amplification toward upstream architectural repair.
Over the past several years— …with increasing clarity in 2024 and early 2025, as spatial and lymph-node–focused analyses have matured—studies have begun to identify active, suppressive control layers operating within tumor-draining lymph nodes themselves. These are not abstract regulatory forces. They are specific cellular actors, spatially localized, and mechanistically defined.
Chief among them are PD-L1–expressing macrophage populations within lymph nodes, positioned at precisely the point where antigen presentation and T-cell fate are decided. Rather than facilitating productive priming, these macrophages blunt it. They attenuate CD8⁺ T-cell activation, constrain proliferative expansion, and—most critically—interfere with the preservation of stem-like progenitor states required for durable immunity. This matters because it resolves a long-standing contradiction in checkpoint biology. Importantly, this suppressive role is spatially and contextually distinct from macrophage functions observed within certain tumor niches, resolving prior confusion without negating earlier observations
Checkpoint inhibitors were designed to remove inhibitory signaling at the tumor site. Yet failure has often occurred even in tumors that clearly express PD-L1, harbor antigenic mutations, and show evidence of immune recognition. The missing explanation was not downstream blockade, but upstream suppression. The emerging LN macrophage axis provides that explanation. While PD-L1⁺ macrophages can exert context-dependent immunostimulatory roles in certain tumor niches, their function within tumor-draining lymph nodes is predominantly suppressive to productive priming.
In this model, immune failure does not originate because PD-1 is irrelevant, nor because the tumor is uniquely evasive. It originates because the immune response was structurally constrained at inception. T cells were instructed under suppressive conditions. Memory was truncated before it could form. What reached the tumor was not a robust, amplifiable population, but a compromised fragment of one. As with most immune regulators, context matters; PD-L1⁺ macrophages may exert stimulatory effects in certain tumor-specific niches, but within tumor-draining lymph nodes their dominant role is suppressive to productive priming.
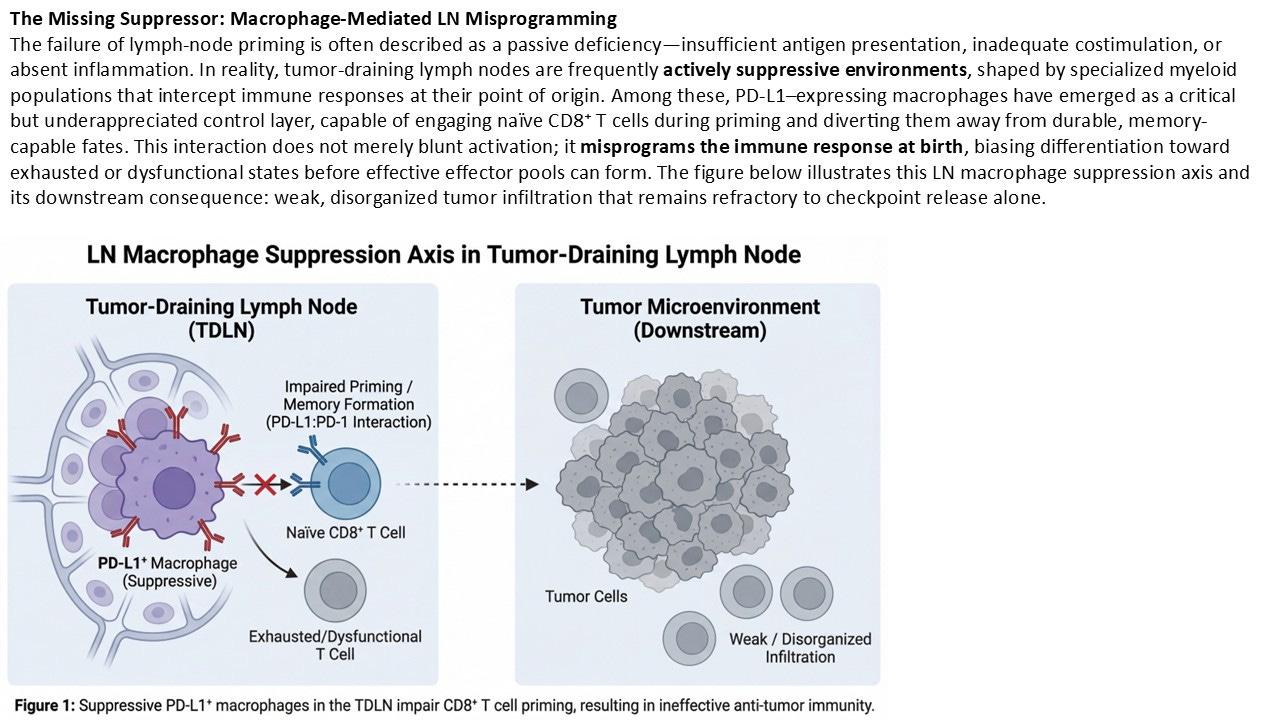
Seen through this lens, the tumor microenvironment becomes less a battlefield and more a downstream checkpoint of a decision already made. Importantly, this LN-localized suppression mirrors what is later observed at the tumor site. Immune-excluded architectures, shallow infiltration, and brittle responses are not independent phenomena. They are spatial echoes of an upstream failure mode. The tumor does not invent the deficit; it inherits it.
This is the point at which LN priming ceases to be speculative. When specific suppressive populations can be identified in lymph nodes, when their molecular signals align with known exhaustion and failure phenotypes, and when their downstream consequences match clinical observation, the debate shifts. We are no longer arguing about whether lymph-node priming might matter. We are confronting evidence that it is a contested control layer, actively shaped by suppressive biology, and decisive for therapeutic outcome. The lymph node is not a passive waypoint. It is an arena. And in many patients, the immune response is losing there long before it ever reaches the tumor.
Immune Lag Is a Structural Feature, Not a False Negative
One of the quiet consequences of impaired lymph-node priming is distorted response timing. When priming is suppressed or mis-programmed, immune responses—if they emerge at all—do so slowly, unevenly, and often outside conventional radiographic expectations. Early tumor enlargement, delayed shrinkage, or prolonged periods of apparent stability have frequently been misclassified as failure, when in fact they reflect a system attempting to rebuild upstream architecture before downstream control can manifest. Consistent with this architecture-first view, recent sequencing studies demonstrate that immunoradiotherapy efficacy depends on intact dendritic migration through tumor-draining lymph nodes, reinforcing priming as a prerequisite rather than a byproduct of response.
This is not an argument for indefinite patience or post-hoc rationalization. It is a recognition that immune assembly precedes immune amplification. iRECIST and related frameworks exist precisely because checkpoint-era biology violated the assumptions of cytotoxic immediacy. LN priming clarifies why. When immune infrastructure must first be repaired—when progenitor pools are reconstituted, trafficking cues re-established, and dendritic licensing restored—time becomes a variable of construction, not indecision. Early non-response, in this context, is not proof of futility. It is often a readout of upstream repair still in progress.
Why This Matters More Than Any Single Biomarker
Once lymph-node priming is recognized as a real, mechanistic control layer—rather than an abstract precondition—the familiar biomarker debates begin to look strangely misaligned. For years, the field has searched for a decisive signal inside the tumor. PD-L1 expression. Tumor mutational burden. Gene signatures. Infiltration scores. Each has promised, at various moments, to sort responders from non-responders with clarity and finality. Each has disappointed.
The reason is not that these biomarkers are useless. It is that they are downstream readouts, interpreted as causes. They describe what the immune response looks like at the tumor endpoint, not whether the response was ever structurally capable of forming. PD-L1 tells us where inhibition is present. TMB tells us how much antigenic opportunity exists. TILs tell us whether immune cells have arrived.
None of them, on their own, tell us whether the immune system successfully navigated the upstream choreography required to generate a durable, amplifiable response. Lymph-node priming does. This is why tumors with high TMB can still fail checkpoint blockade. Antigen alone does not guarantee effective priming. It must be processed, presented, and coupled to a lymph-node environment that favors progenitor preservation over terminal exhaustion. It is why PD-L1–positive tumors can remain inert. Removing an inhibitory signal at the tumor site cannot compensate for suppressive instruction that occurred earlier, in the lymph node, before effector populations were ever formed.
And it is why eTILs—particularly their spatial patterning—consistently outperform single-axis biomarkers when durability is examined rather than initial shrinkage. They do not function as magic predictors. They function as structural receipts, confirming whether the upstream system worked. Once this hierarchy is restored, many apparent contradictions dissolve.
Checkpoint inhibitors do not “fail” because they are poorly designed. They fail because amplification presupposes architecture. Where that architecture is intact, even modest checkpoint intervention can unlock durable benefit. Where it is compromised, escalation rarely helps and often harms. This reframing also explains why attempts to stack biomarkers have produced diminishing returns. Combining multiple downstream readouts does not reconstruct a collapsed upstream process. It merely adds resolution to the observation of failure.
Lymph-node priming, by contrast, does not compete with existing biomarkers. It orders them. It explains why TMB modifies probability but does not rescue immune deserts; why PD-L1 expression can be necessary but insufficient; why infiltration without organization predicts fragility rather than strength. In this hierarchy, LN priming is not another variable to be optimized. It is the load-bearing layer upon which all others depend. And once that layer is acknowledged, the question facing immunotherapy shifts. It is no longer “which biomarker best predicts response?” It becomes “what must be repaired before response is even possible?”
Repair vs Escalation: The Strategic Consequence
Once lymph-node priming is understood as a structural prerequisite rather than a peripheral curiosity, a quiet but profound consequence follows: many immunotherapy failures are not failures of intensity, but failures of construction. For years, the dominant response to checkpoint resistance has been escalation. More checkpoints. Higher doses. Earlier lines. Combinations layered atop combinations. The implicit assumption has been that immune resistance reflects insufficient pressure—that if the brakes are removed harder, longer, or earlier, the system will eventually respond.
But escalation presumes that something functional already exists to be amplified. If LN priming has failed—if progenitor populations were never preserved, if instruction occurred under suppressive conditions, if the immune response was malformed at inception—then escalation becomes a blunt instrument. It increases toxicity, accelerates exhaustion, and narrows therapeutic windows without restoring the missing architecture.
This is why so many aggressive combinations produce impressive immune activation markers alongside disappointing durability. They generate motion without structure. Repair, by contrast, begins from a different premise. It asks not how forcefully to push the immune system, but what foundational elements are absent or impaired. It recognizes that before amplification can matter, the immune response must be rebuilt in the right place, in the right sequence, and under the right signaling conditions.
This distinction has implications not only for therapy selection, but for trial design itself — including endpoints, timing expectations, and the interpretation of early non-response. This is not semantic reframing. It has direct clinical consequences. Repair-oriented strategies aim to restore lymph-node competence: to re-enable productive antigen presentation, to rebalance cytokine signaling in favor of memory-preserving pathways, and to reconstitute the progenitor pools from which durable responses are drawn. Only after this substrate is restored does checkpoint amplification regain relevance.
Seen this way, escalation without repair is not merely inefficient—it is misaligned. It treats late-stage failure as though it were an early-stage deficiency. This distinction also clarifies why some patients exhibit delayed or atypical response kinetics. In repair-first paradigms, time is not spent shrinking tumors immediately, but rebuilding immune infrastructure. When responses emerge, they do so from a fundamentally different starting point, often with greater durability and resilience.
The strategic implication is unavoidable: not all non-responders should be escalated. Some must first be repaired. And until development strategies reflect that distinction, the field will continue to mistake architectural failure for therapeutic inadequacy.
Why MDNA11 Fits the Architecture: β-Bias as Repair, Not Escalation
Once immune failure is understood as architectural rather than energetic, the question is no longer which therapy is strongest, but which therapy operates at the contested layer.
This is where β-biased IL-2 signaling becomes conceptually distinct — not because it is more inflammatory, but because it is positioned correctly.
MDNA11’s β-selective design preferentially engages CD8⁺ T cells and NK cells while avoiding the α-chain–mediated expansion of regulatory populations that can reinforce suppression. But the more important distinction is where that signaling is functionally relevant. Delivered systemically and rhythmically, β-biased IL-2 reinforces STAT5 signaling in LN-resident TCF1⁺ progenitor CD8⁺ cells, precisely the population whose preservation determines whether immune responses can be renewed rather than consumed.
This matters even more in light of emerging neuroimmune and stromal governance data. The Cell study describing inter-organ neural circuits adds a critical layer above LN priming: immune assembly can be actively permissioned or throttled by systemic signals unrelated to antigen or checkpoint engagement. In that context, downstream activation strategies risk forcing effector differentiation in an environment where renewal is still constrained.
β-biased IL-2 does not attempt to overpower that constraint. Instead, it stabilizes the renewal layer beneath it.
By reinforcing rhythmic STAT5 signaling — rather than inducing continuous supraphysiologic activation — MDNA11 supports progenitor maintenance under conditions where immune competence exists but is contested. This helps explain why β-biased IL-2 can restore productive immunity without forcing terminal differentiation, excessive cytokine release, or premature exhaustion.
In architectural terms, MDNA11 does not bypass upstream governance. It repairs immune assembly so that governance can be meaningfully re-engaged downstream. That is a fundamentally different posture from bispecific activation, intratumoral cytokine delivery, or escalation-based checkpoint stacking — all of which assume that the assembly floor is already intact.
This is also why MDNA11’s relevance does not depend on being “better” than other modalities. It depends on whether the modality operates above or below the immune assembly threshold. Under that lens, β-biased IL-2 is not an amplifier competing for downstream effect — it is a repair signal restoring the conditions under which amplification can work at all.
Why the “Speculative” Label Persisted—and Why It Will Fade
If lymph-node priming explains so much—if it resolves contradictions that have frustrated immunotherapy for years—then an obvious question follows: why did it take so long to be taken seriously? The answer is not a lack of data. It is a mismatch between how the immune system actually works and how the field learned to observe it.
Modern oncology has been built around what is easiest to see. Tumors can be biopsied. PD-L1 can be stained. Mutations can be counted. Responses can be measured on scans. The lymph node, by contrast, sits upstream, dynamic, and largely inaccessible. It does its most important work early, transiently, and out of view. As a result, the field grew accustomed to reasoning backward—from tumor outcomes to presumed causes. When responses failed, explanations clustered around what could be measured at the endpoint. LN priming, which resists static snapshots and binary classification, did not fit cleanly into that paradigm.
There was also a deeper conceptual resistance. LN priming does not lend itself to a single biomarker, a single cutoff, or a single intervention. It is architectural, sequential, and contingent. It requires thinking in systems rather than switches. For a field eager for decisive predictors and fast answers, that complexity was easy to label as speculative. But the ground has shifted.
Advances in spatial biology, single-cell profiling, and translational pattern recognition have begun to make the invisible visible. eTIL patterning now reads as a downstream signature of upstream competence. LN-localized suppressive macrophage populations provide mechanistic anchors where theory once stood. Durability, not just response, has become the metric that matters. Most importantly, the clinical failures have accumulated. The limits of escalation are no longer abstract. They are written into trial after trial where intensity increased and benefit did not. In that environment, explanations that once seemed “theoretical” begin to look simply unexamined.
The speculative label persists longest not where ideas are weakest, but where they are hardest to operationalize. LN priming has required the field to look upstream, to slow down, and to accept that some failures cannot be fixed by pressing harder on downstream levers. That reluctance is fading—not because the idea has changed, but because the evidence has become impossible to ignore.
Lymph-node priming is no longer an argument from plausibility. It is an argument from structure, from mechanism, and from convergence. As the tools to observe it mature, and as strategies shift from escalation toward repair, what once felt speculative will come to seem obvious in hindsight. Not because the field suddenly became wiser—but because it finally learned where to look.
From Discovery to Architecture: Why Lymph-Node Priming Is the Unifying Constraint
In 2022, Huang et al. published a quietly consequential Cell paper that, in retrospect, resolved a central ambiguity in checkpoint biology. Their work demonstrated that the bona fide responders to PD-1/PD-L1 blockade are not terminally exhausted, tumor-resident T cells, but a distinct population of tumor-draining lymph node–derived, TCF1⁺ tumor-specific memory CD8⁺ T cells. These cells reside upstream of the tumor, retain self-renewal capacity, avoid exhaustion-associated epigenetic scarring, and serve as the renewable source of effector waves that ultimately traffic into the tumor microenvironment.
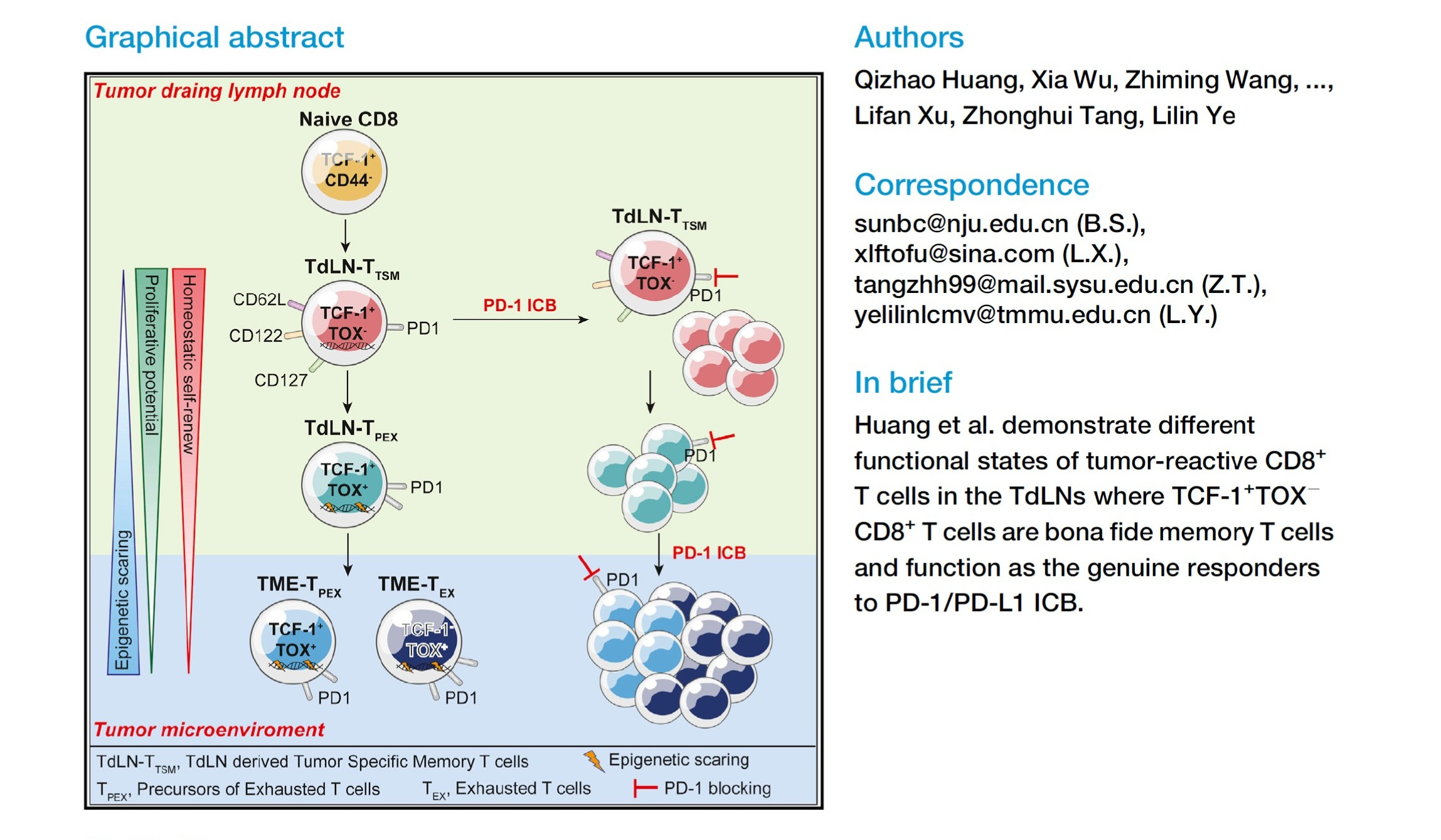
The discovery itself was correct. What failed to propagate was the framework. The terminology introduced in that paper — TdLN-T<sub>TSM</sub> — never meaningfully entered clinical or translational discourse. Instead, the field adopted a looser shorthand around “TCF1⁺ progenitors,” often abstracted away from their anatomical origin, renewal dynamics, and systems-level implications. As a result, the biological insight remained fragmented: appreciated mechanistically, but rarely operationalized.
PRISM-11 reframes that same biology not as a cell-type curiosity, but as a hard architectural constraint on durable immunotherapy. The updated framework presented here translates the original discovery into a modern immuno-oncology architecture with three explicit layers:
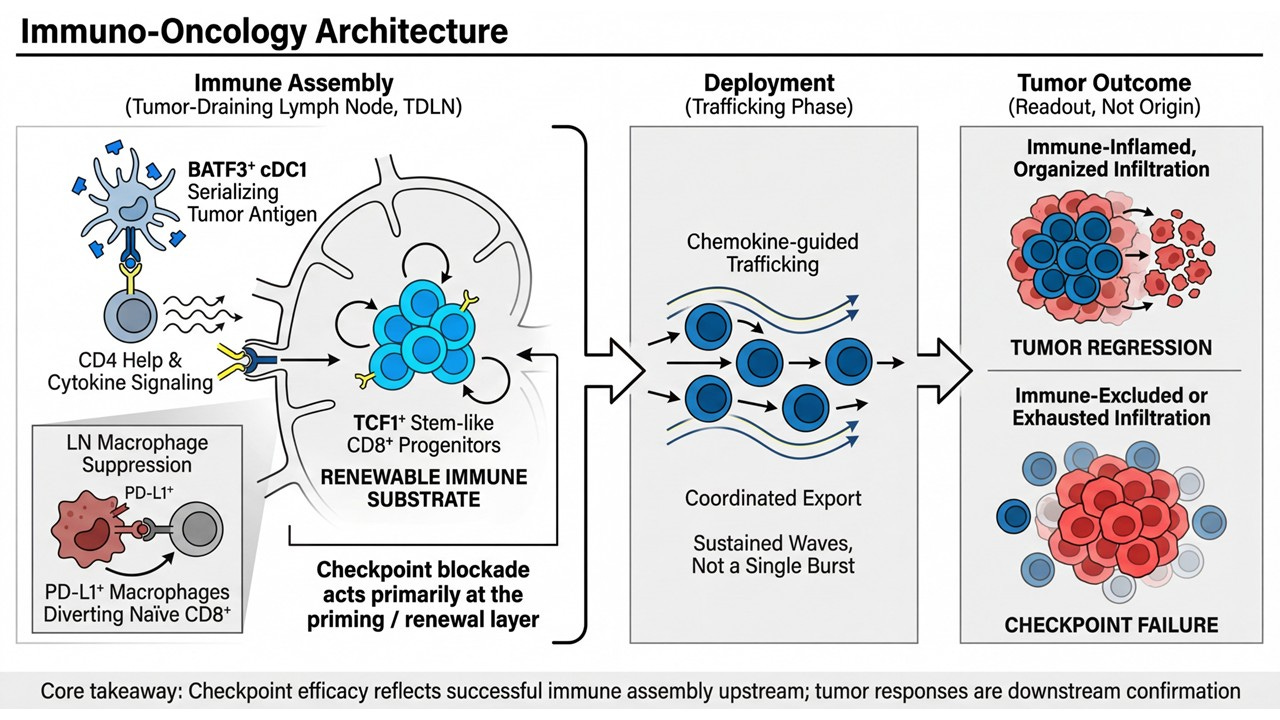
Immune Assembly (Tumor-Draining Lymph Node):
BATF3⁺ cDC1–mediated antigen serialization, CD4 help, and β/γ cytokine signaling converge to maintain a renewable pool of LN-resident TCF1⁺ progenitor CD8⁺ T cells. This is where immune potential is built or lost.Deployment (Trafficking Phase):
Coordinated, chemokine-guided export of effector progeny occurs in sustained waves, not single bursts — a direct function of upstream progenitor renewal.Tumor Outcome (Readout, Not Origin):
Organized infiltration and tumor regression reflect successful upstream assembly; immune-excluded or exhausted phenotypes reflect its failure. The tumor is a downstream sensor, not the command center.
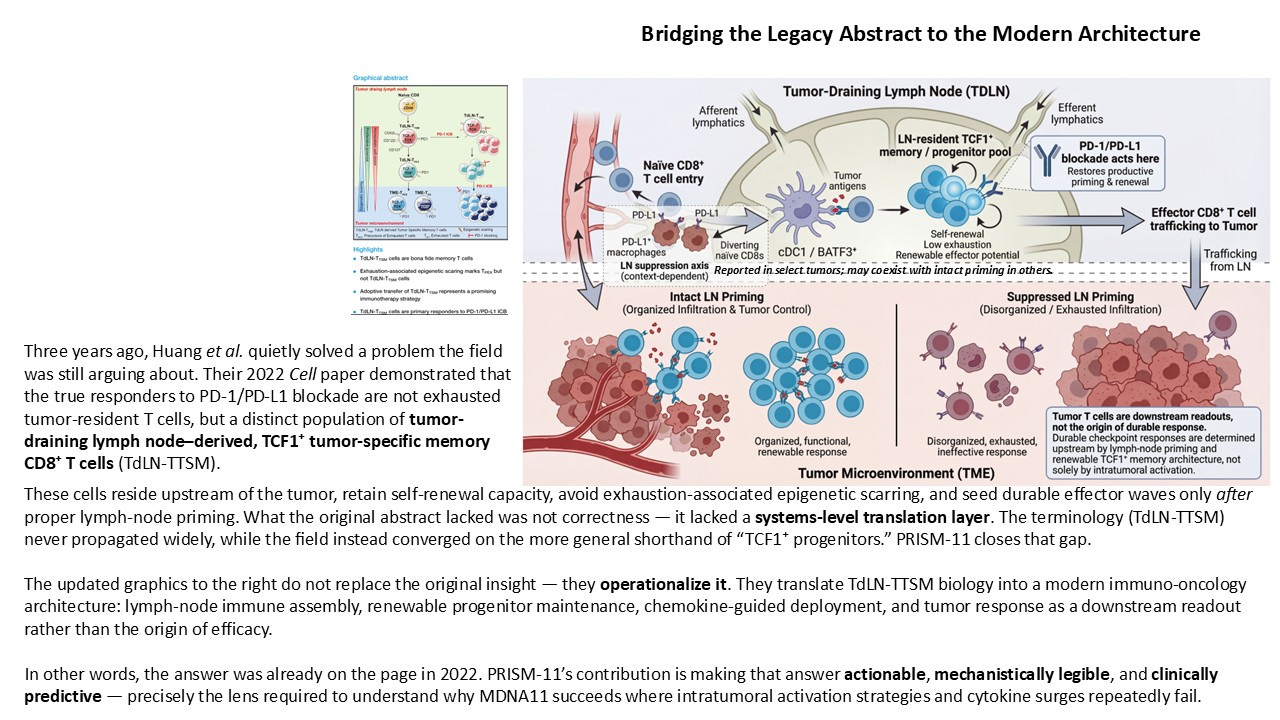
Within this architecture, checkpoint blockade acts primarily at the priming and renewal layer, not by reinvigorating terminally exhausted cells in situ, but by restoring productive differentiation and export from the lymph node. This reconciles years of clinical paradoxes — delayed responses, pseudoprogression, cfDNA-radiology mismatches — that make little sense if the tumor is treated as the origin of efficacy.
This reframing matters because it collapses an entire competitive landscape into a single question: does the therapy rebuild lymph-node immune assembly and renewable progenitor architecture — or does it act downstream of that constraint?
Under this lens, distinctions between modalities become secondary:
IL-2 vs IL-15 is less important than whether signaling is positioned to support LN-resident progenitor maintenance.
WTX-124 enables tumor-conditional cytokine release, but still operates downstream of lymph-node assembly.
IBI-363 and other bispecifics provide localized activation without guaranteed renewal of the progenitor pool.
Intratumoral IL-2, vaccines, and oncolytics can inflame the tumor without generating system-level immune memory.
Checkpoint blockade alone releases inhibitory brakes but cannot rebuild a depleted engine.
This is why the claim advanced here is not that one drug is “better” than another. The claim is more fundamental: durable immunotherapy only works if it rebuilds lymph-node immune assembly and renewable progenitor architecture. Everything else is downstream noise.
Within that constraint, MDNA11 is not positioned as an exception, but as an example of alignment by design — a systemically delivered, β/γ-selective cytokine engineered to engage the lymph-node priming axis rather than bypass it. Its relevance follows from the architecture, not the other way around. The answer, in hindsight, was already present in 2022. PRISM-11’s contribution is showing why it matters — and why every successful immunotherapy, regardless of class, must ultimately obey the same upstream rules.
The Quiet Conclusion
Lymph-node priming was never speculative because it lacked evidence. It was speculative because it required the field to look upstream—to think architecturally rather than reactively, to distinguish construction from amplification, and to accept that no amount of downstream activation can compensate for a system that was never properly assembled.
For more than a decade, immuno-oncology optimized around what was visible: tumor biopsies, intratumoral T cells, checkpoint expression, response rates. The tumor became the focal plane, and success or failure was inferred from what could be sampled most easily. But tumors are not where durable immunity is built. They are where it is tested. When responses proved transient, heterogeneous, or inexplicable, the answer was assumed to lie in better targets, stronger agonism, tighter delivery, or deeper combinations—more force applied to the same downstream layer.
What has changed is not the biology, but our ability to see it. Spatial biology, single-cell lineage tracing, lymph-node–aware trial designs, and durability-weighted outcomes have converged to illuminate the upstream layer that was always doing the real work. Tumor-draining lymph nodes are not passive conduits; they are the immune system’s assembly floor. It is there that antigens are serialized, progenitor pools are renewed, exhaustion is avoided or imposed, and memory architecture is either preserved or lost. When that structure is intact, downstream biomarkers align and responses endure. When it is suppressed or bypassed, no degree of intratumoral activation can reliably rescue the system.
This reframes checkpoint failure in a fundamental way. Checkpoint inhibitors have not failed because immunity is absent, nor because tumors are uniformly non-immunogenic. They fail, in many patients, because the immune response was never structurally assembled in a renewable way. PD-1 blockade releases brakes, but it does not rebuild engines. Without lymph-node priming and progenitor renewal, the system accelerates briefly—and then stalls.
Importantly, lymph-node priming does not compete with existing biomarkers or therapeutic classes; it orders them. Tumor mutational burden, PD-L1 expression, TIL density, cytokine bursts, bispecific engagement, conditional cytokine delivery, vaccines, and even cell therapies all operate downstream of this assembly layer. Their variability, their flashes of efficacy, and their frequent failures become intelligible once viewed through the lens of upstream immune architecture. What appeared contradictory becomes coherent. What seemed noisy resolves into pattern.
Seen this way, the central question of durable immunotherapy is no longer which modality is superior, but whether a given approach rebuilds—or depends upon—lymph-node immune assembly and a renewable progenitor substrate. Everything else is execution detail.
In that sense, lymph-node priming is not a new mechanism competing for attention. It is the unifying constraint that quietly governs them all. And once that constraint is acknowledged, much of what has puzzled immuno-oncology for the past decade begins, finally, to make sense.
References
Chamoto K, et al. Lymph node CD8⁺ T cell priming determines the efficacy of immune checkpoint blockade.
Nature Immunology (2020).
https://www.nature.com/articles/s41590-020-0658-3Im SJ, et al. Defining CD8⁺ T cells that provide the proliferative burst after PD-1 therapy.
Nature (2016).
https://www.nature.com/articles/nature19330Miller BC, et al. Subsets of exhausted CD8⁺ T cells differentially mediate tumor control and respond to checkpoint blockade.
Nature Immunology (2019).
https://www.nature.com/articles/s41590-019-0412-1Sade-Feldman M, et al. Defining T cell states associated with response to checkpoint immunotherapy in melanoma.
Cell (2018).
https://www.cell.com/cell/fulltext/S0092-8674(18)30610-1Yost KE, et al. Clonal replacement of tumor-specific T cells following PD-1 blockade.
Nature Medicine (2019).
https://www.nature.com/articles/s41591-019-0522-3Spitzer MH, et al. Systemic immunity is required for effective cancer immunotherapy.
Cell (2017).
https://www.cell.com/cell/fulltext/S0092-8674(17)30787-8Binnewies M, et al. Understanding the tumor immune microenvironment (TIME) for effective therapy.
Nature Medicine (2018).
https://www.nature.com/articles/s41591-018-0014-xKeren L, et al. A structured tumor-immune microenvironment is associated with better prognosis in triple-negative breast cancer.
Cell (2018).
https://www.cell.com/cell/fulltext/S0092-8674(18)30344-4Saltz J, et al. Spatial organization and molecular correlation of tumor-infiltrating lymphocytes using deep learning on pathology images.
Cell Reports (2018).
https://www.cell.com/cell-reports/fulltext/S2211-1247(18)30659-0Galon J, et al. Towards the introduction of the ‘Immunoscore’ in the classification of malignant tumours.
Journal of Pathology (2014).
https://onlinelibrary.wiley.com/doi/10.1002/path.4287Fridman WH, et al. The immune contexture in cancer prognosis and treatment.
Nature Reviews Clinical Oncology (2017).
https://www.nature.com/articles/nrclinonc.2017.101Dammeijer F, et al. Tumor-educated macrophages in lymph nodes suppress antitumor immunity.
Nature Communications (2020).
https://www.nature.com/articles/s41467-020-15840-1Broz ML, et al.
Dissecting the tumor myeloid compartment reveals rare activating antigen-presenting cells critical for T cell immunity.
Cancer Immunology Research (2014)
https://aacrjournals.org/cancerimmunolres/article/2/9/830/468557Salmon H, et al. Expansion and activation of CD103⁺ dendritic cell progenitors at the tumor site enhances tumor responses to therapeutic PD-L1 and BRAF inhibition.
Immunity (2016).
https://www.cell.com/immunity/fulltext/S1074-7613(16)30130-4Garris CS, et al. Successful anti–PD-1 cancer immunotherapy requires T cell–dendritic cell crosstalk involving the cytokines IFN-γ and IL-12.
Immunity (2018).
Successful anti-PD-1 cancer immun... | Archive ouverte UNIGEChen DS & Mellman I. Elements of cancer immunity and the cancer–immune set point.
Nature (2017).
https://www.nature.com/articles/nature21349Topalian SL, et al. Mechanism-driven biomarkers to guide immune checkpoint blockade in cancer therapy.
Nature Reviews Cancer (2016).
https://www.nature.com/articles/nrc.2016.36Zilionis R, et al. Single-cell transcriptomics of human and mouse lung cancers reveals conserved myeloid populations across individuals and species. Immunity (2019)
https://www.cell.com/immunity/fulltext/S1074-7613(19)30057-4Wu SZ, et al. A single-cell and spatially resolved atlas of human breast cancers.
Nature Genetics (2021)
https://www.nature.com/articles/s41588-021-00830-4van der Leun AM, et al. Tumor-draining lymph nodes as multi-omics hubs in cancer immunotherapy. Frontiers in Immunology (2025).
https://www.frontiersin.org/articles/10.3389/fimmu.2025.1298437 Review positioning TDLNs as integrative, multi-omics immune control centers; directly supports observability and non-speculative framing.Wang Y, et al. Effective sequencing of immunoradiotherapy requires dendritic cell migration through tumor-draining lymph nodes.
Nature Communications (2025).
https://www.nature.com/articles/s41467-025-47291-3 Demonstrates LN-dependent DC trafficking as a prerequisite for combined modality efficacy; supports timing and repair-before-amplification logic.Tran E, et al. Lymph-node–targeted KRAS vaccine induces systemic antitumor immunity in patients with solid tumors.
Nature Medicine (2025).
https://www.nature.com/articles/s41591-025-02841-9 Phase 1 clinical study explicitly targeting lymph-node immune programming; supports the 2024–2025 “intervention-level” shift.Dammeijer F, et al. Tumor-draining lymph nodes: friend or foe in immune checkpoint blockade?
Trends in Cancer (2025).
https://www.cell.com/trends/cancer/fulltext/S2405-8033(25)00041-7 Balanced review explaining historical ambiguity; supports your argument that conceptual confusion ≠ biological uncertainty.Gubin MM, et al. Context-dependent roles of PD-L1⁺ macrophages in shaping antitumor immunity.
Cell Reports Medicine (2024).
https://www.cell.com/cell-reports-medicine/fulltext/S2666-3791(24)00086-4 Provides nuance on PD-L1⁺ macrophages while preserving the dominant suppressive role in tumor-draining lymph nodes.
25. Huang et al., Cell, 2022 The primordial differentiation of tumor-specific memory CD8⁺ T cells as bona fide responders to PD-1/PD-L1 blockade in draining lymph nodes
Cell (2022)
26. Chamoto et al., Nature Immunology, 2020 Lymph node CD8⁺ T cell priming determines the efficacy of immune checkpoint blockade
Nature Immunology (2020)
27. Im et al., Nature, 2016 Defining CD8⁺ T cells that provide the proliferative burst after PD-1 therapy Nature (2016)
28. Siddiqui et al., Nature Immunology, 2019 Intratumoral Tcf1⁺PD-1⁺ CD8 T cells sustain anti-tumor immunity Nat Immunol (2019)
29. Yost et al., Nature Medicine, 2019 Clonal replacement of tumor-specific T cells following PD-1 blockade Nat Med (2019)
30. Dammeijer et al., Nature Communications, 2020 Tumor-educated macrophages in lymph nodes suppress anti-tumor immunity Nat Commun (2020)
31. Broz et al., Cancer Immunology Research, 2015 Dissecting the tumor myeloid compartment reveals rare activating APCs critical for T cell immunity
32. Spitzer et al., Cell, 2017 Systemic immunity is required for effective cancer immunotherapy
33. Trends in Cancer, 2024–2025 (review) Tumor-draining lymph nodes as dual regulators of immunity and metastasis
34. Frontiers in Immunology, 2025 (review) Multi-omics mapping of tumor-draining lymph nodes reveals immune fate decisions
35. Chen & Mellman, Nature, 2017 Elements of cancer immunity and the cancer–immune set point
MDNA-11 is positioned to be a primary enabling therapy in modern immuno-oncology!!!!