
Operationalizing Layer 2: Sub-Layer Stratification of Lymph Node Governance
From Rhythmic Renewal to Conditional Restoration — Reading the 2026 MDNA11 Signals

Author’s Note:
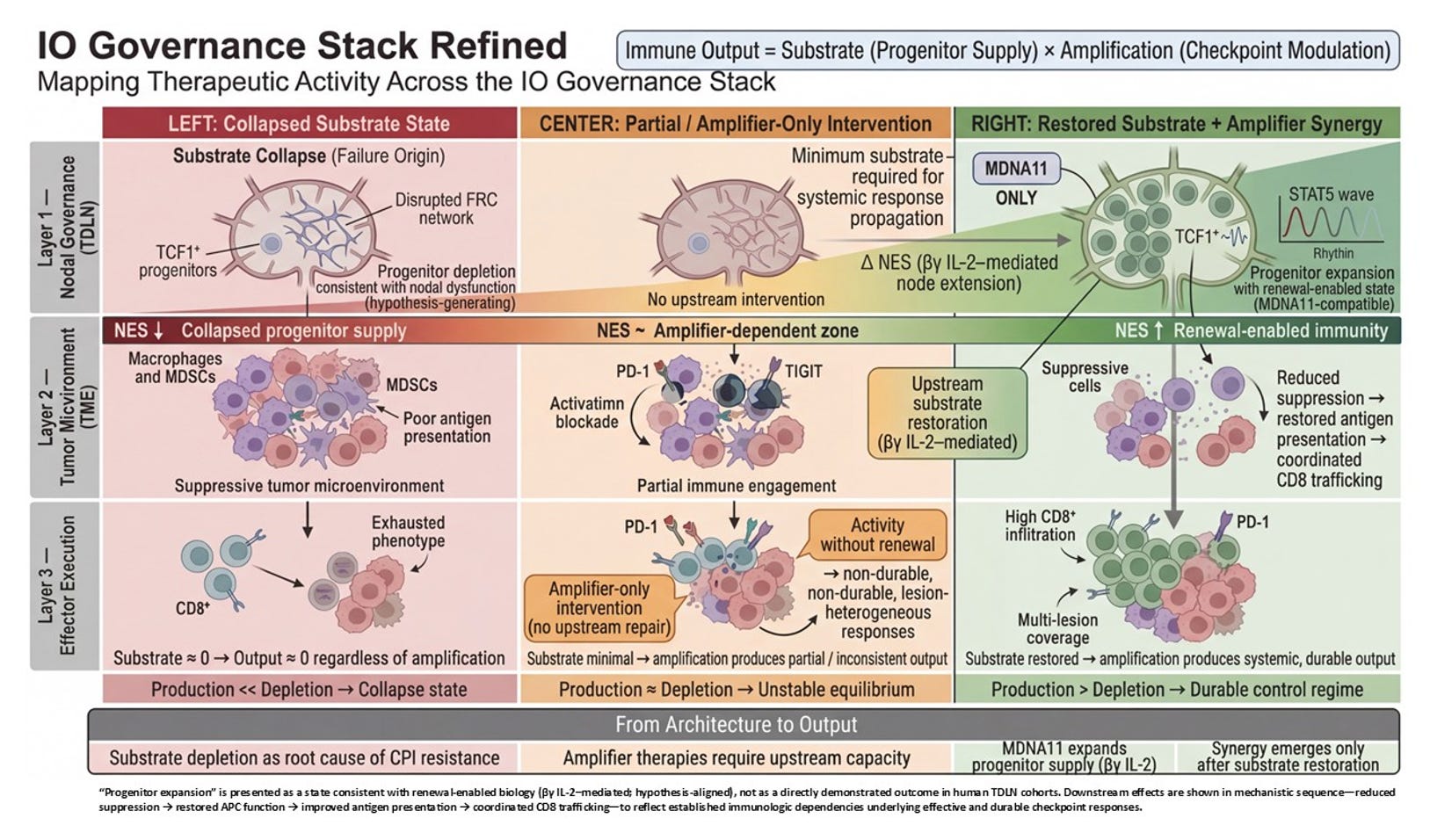
This analysis operationalizes the Layer 2 governance architecture first mapped in “The Governance Organ.” Where that framework described the collapse stack and restoration windows, the present work resolves the tumor-draining lymph node into its discrete mechanical and biological sublayers—from afferent ingress through the epigenetic boundary—and identifies, directionally, where MDNA11 intervenes.
The February 2026 corporate deck supplies the first clinical contour lines. Patient selection by line-of-therapy functions as an architectural proxy. Monotherapy activity in selected cohorts is consistent with substrate-level activity prior to execution-layer amplification. Combination depth is broadly consistent with proper sequencing: restoration first, execution second.
Ten months ago, “A Billion-Dollar Layup in Salvage Therapy” argued that even a conservative salvage path justified a multi-billion-dollar valuation floor. Layer 2 stratification, combined with the new data cut, does not revise that thesis—it hardens it. By making conditional restoration measurable and potentially approvable, the framework transforms salvage from a fallback into a structurally defensible foundation. Durability does not live in peak activation. It lives in substrate integrity. This piece makes that distinction operational.
I recognize that there may be frustration with discussions of TAM, valuation, and long-range potential given the current state of the equity. That reaction is understandable—and not new territory for long-time shareholders. There is an uncomfortable reality sitting underneath this discussion that is worth acknowledging directly. At roughly $50–60 million USD of market value, the current tape is not pricing a salvage platform (or any platform for that matter), a mono or combination strategy, or even a coherent clinical signal. It is pricing uncertainty, financing risk, and the possibility that development-stage biology fails to translate into durable outcomes. It is also, in part, a function of market structure—thin liquidity, borrow availability in an OTC setting (Fidelity currently showing ~10.6%), and the types of pressures that tend to concentrate in small-cap biotech when capital is scarce.
For holders, that dislocation is not abstract. It is felt daily—in the chart, in the volatility, and in the constant question of whether the system will recognize value before, during, or after a financing event, or whether time itself becomes the dominant cost. Nothing in this work dismisses that reality. If anything, it is written because of it. The purpose of the framework is not to argue that the market is “wrong” in a simplistic sense, or that value must immediately converge. Markets can remain dislocated for extended periods, particularly in capital-constrained environments. The purpose is narrower and more structural: to determine whether, beneath the noise and high-level summaries of clinical outcomes, the underlying biology and signal support a valuation floor that is materially different from what is currently implied—and to make the “how” and “why” of that assessment explicit.
In that context, the relevant question is not simply why the stock is down. It is whether the assumptions used to construct the original salvage thesis were themselves too restrictive—even under deliberately conservative conditions.
Regeneration or Exhaustion
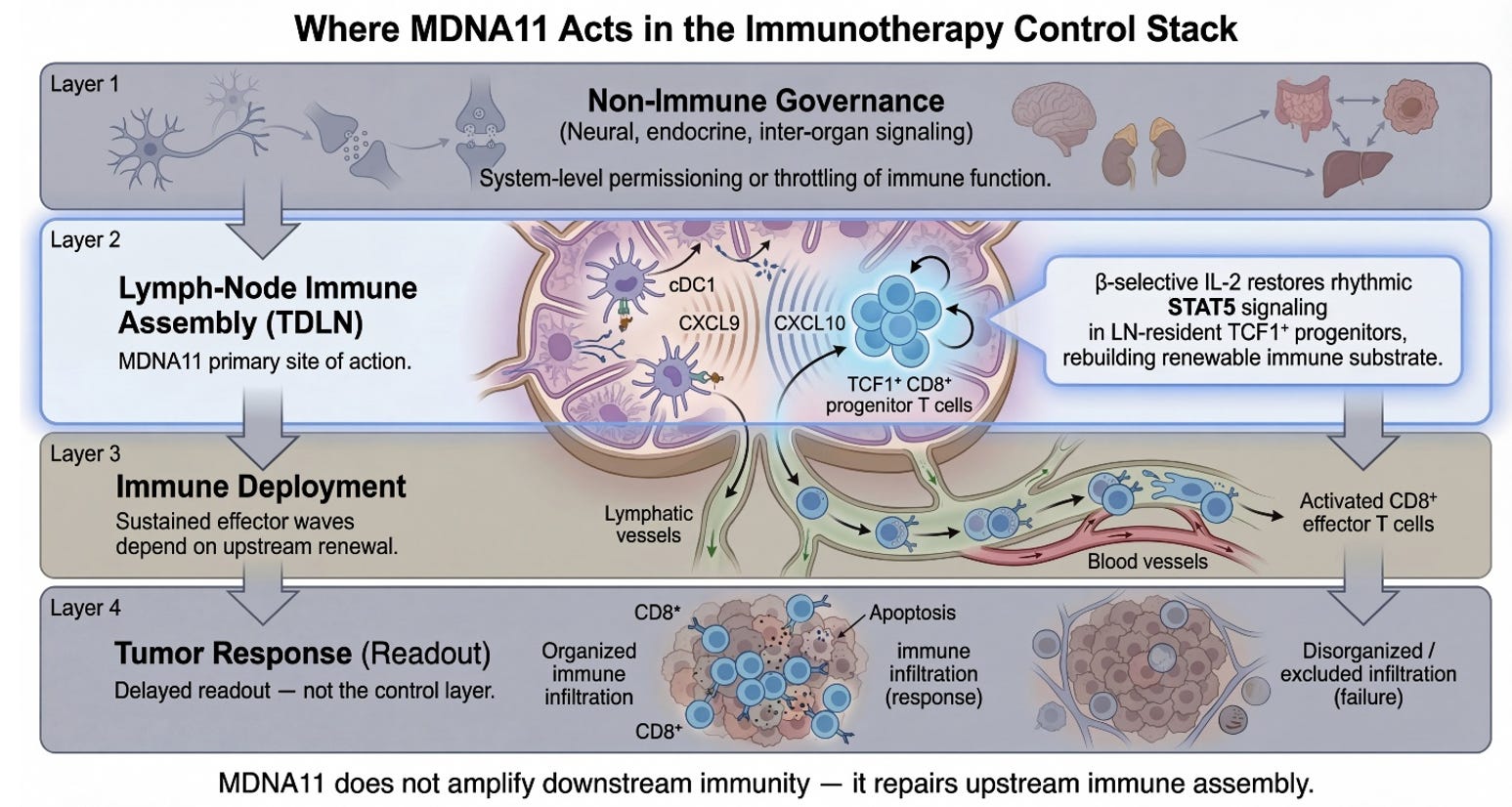
Layer 2 is where the immune system decides whether it is capable of sustaining war. Not whether it can fire once. Not whether it can briefly infiltrate. But whether it can renew itself under pressure. The stratification of Layer 2 is not a stylistic elaboration of the control stack; it is the architectural cross-section of immune durability. It takes what is often treated as a black box—“lymph node priming”—and resolves it into the discrete mechanical and biological sublayers that determine whether immune competence can regenerate or collapses into exhaustion.
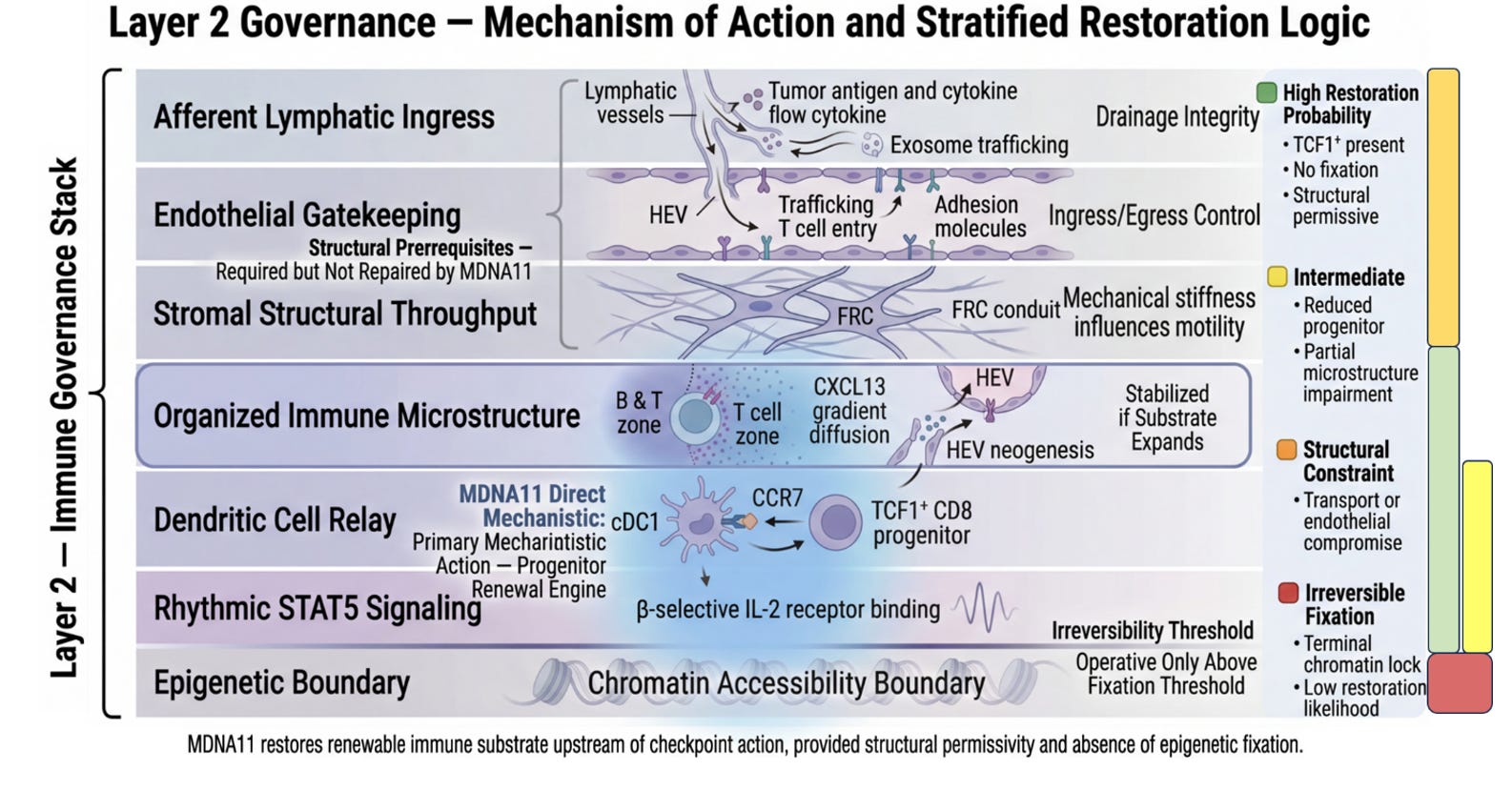
At the top of the Layer 2 graphic sits Afferent Lymphatic Ingress. This is the drainage architecture. Tumor antigens, cytokines, and vesicular debris must physically reach the tumor-draining lymph node. Without intact lymphatic vessels, without preserved flow, there is no informational feed into the immune assembly site. This layer is not glamorous, but it is foundational. It determines whether the node even knows the tumor exists. MDNA11 does not repair broken drainage. It presupposes that antigen can arrive. This is a structural prerequisite, not a therapeutic target.
Beneath ingress lies Endothelial Gatekeeping. High endothelial venules regulate who may enter and who may not. Adhesion molecules, trafficking cues, and chemokine gradients determine whether naïve and memory T cells can access the node and participate in priming. If ingress is about information arriving, gatekeeping is about participants being allowed into the assembly hall. Again, MDNA11 does not reengineer HEVs. It operates within their permissions. If endothelial trafficking is severely compromised, substrate restoration downstream will not translate into deployment.
Next comes Stromal Structural Throughput. Fibroblastic reticular cell networks create the physical conduit system of the node. They determine how signals diffuse, how dendritic cells migrate, how T cells scan antigen. Mechanical stiffness, conduit integrity, and matrix organization influence motility and encounter probability. This is the node’s internal plumbing. Without it, signals dissipate chaotically. MDNA11 does not rebuild FRC scaffolds. It assumes the architecture is sufficiently intact to support renewal once signaling resumes.
These three layers together form what the slide correctly labels Structural Prerequisites. They define permissivity. They are required but not repaired by MDNA11. That distinction matters. It draws a line between transport failure and assembly failure. Many immunotherapy disappointments are transport problems masquerading as immune incompetence.
Below the structural layers sits Organized Immune Microstructure. This is where architecture becomes adaptive. B and T cell zones segregate. CXCL13 gradients form. HEV neogenesis may occur in response to chronic antigenic stimulation. Tertiary lymphoid features may emerge or collapse. Microstructure determines whether priming is spatially organized or diffusely inefficient. It is here that sustained competence is either stabilized or eroded over time. MDNA11 does not directly induce tertiary lymphoid structures. But if progenitor substrate expands beneath this layer, microstructure can stabilize rather than deteriorate. That is why the green zone in your stratification mapping includes this layer: not because MDNA11 builds it de novo, but because substrate renewal supports its persistence.
Beneath microstructure lies the Dendritic Cell Relay. This is the interface between antigen presentation and progenitor fate. cDC1 cells process tumor antigen, present it in the context of costimulatory cues, and engage TCF1⁺ CD8 progenitors. CCR7 signaling maintains migratory coherence. If this relay collapses, progenitors cannot be refreshed. This is one of the two layers where MDNA11’s influence becomes direct. “By engaging β-selective IL-2 receptors, MDNA11 delivers intermittent STAT5 signaling consistent with progenitor-compatible renewal kinetics observed in preclinical systems. It does not replace antigen presentation; it enables progenitors to respond to it.
Immediately below sits Rhythmic STAT5 Signaling. This is the renewal engine. The distinction between tonic overstimulation and rhythmic reinforcement is not cosmetic; it is fate-defining. Sustained high-intensity IL-2 exposure can drive terminal differentiation or exhaustion. Rhythmic STAT5 activation within progenitor populations maintains self-renewal while allowing downstream effector differentiation to proceed in waves. This is the narrowest and most precise locus of MDNA11’s mechanistic claim. It expands renewable substrate rather than amplifying terminal effectors. It acts upstream of checkpoint blockade, not in parallel with it.
Finally, the Epigenetic Boundary marks the irreversibility threshold. Once chromatin accessibility collapses into fixed exhaustion states, transcriptional programs become locked. Progenitor identity cannot be reconstituted by cytokine signaling alone. This is the red line in your mapping. MDNA11 operates only above this boundary. It cannot unlock terminal fixation. That restraint is not a weakness; it is a definition of scope. It allows you to articulate where the molecule will not work.
Taken together, the stratification of Layer 2 transforms “lymph node priming” into its own governance stack. Transport. Gatekeeping. Structural throughput. Microstructure. Relay. Renewal signaling. Epigenetic boundary. Each layer defines a distinct failure mode. Each defines a distinct inclusion or exclusion logic for trial design. And critically, only two of those layers are directly modulated by MDNA11.
What the slide represents, therefore, is not merely where MDNA11 acts. It represents conditional efficacy. Green is not optimism; it is architectural coherence: substrate present, structure permissive, no fixation. Yellow is substrate scarcity. Orange is transport or endothelial constraint outside MDNA11’s repair domain. Red is terminal chromatin lock. That stratification logic is embedded in the architecture itself.
In contrast to many immuno-oncology combinations that cannot disentangle which layer they are affecting, this framework specifies the intervention plane. Checkpoint inhibitors unlock execution. Bispecifics redirect killing. Tumor-activated cytokines intensify local inflammation. MDNA11 is designed to expand renewable substrate upstream of execution-layer amplifiers. It is not an amplifier. It is a renewal engine.
The significance of this becomes clearest when considering regulatory dialogue. When asked why MDNA11 may succeed where PD-1 failed, the answer is no longer “stronger activation.” It is that prior failure may have reflected substrate depletion rather than checkpoint dominance. If TCF1⁺ progenitors are diminished but not epigenetically fixed, renewal can be restored. If fixation has occurred, futility should be anticipated. That is a mechanistic inclusion criterion, not an empirical hope.
The Layer 2 graphic slide above, therefore, is not ornamental detail. It is a map of immune durability. It clarifies what must be intact, what can be repaired, and what lies beyond recovery. And within that map, MDNA11 occupies a narrow but strategically powerful position: the restoration of rhythmic STAT5-mediated progenitor renewal within a structurally permissive lymph node architecture. It does not promise universal rescue. It promises conditional restoration. That conditionality is not a limitation; it is what makes the architecture defensible.
“That architectural precision now finds its first clinical contour lines in the February 2026 corporate deck, where line-of-therapy segmentation and response patterns begin to map directly onto the Layer 2 governance zones — turning a mechanistic model into an operational filter.
From Architecture to Waterfall: Clinical Echoes of Layer 2 Stratification
Layer 2 was never meant to be decorative theory. It was written as a structural claim about where durability lives and where it dies. The blow-out of the tumor-draining lymph node was not an aesthetic flourish—it was an attempt to make visible the invisible governance organ that decides whether immune war can be sustained. Transport. Gatekeeping. Stromal throughput. Microstructure. Dendritic relay. Rhythmic STAT5 renewal. Epigenetic boundary. Each sublayer exists because collapse does not occur all at once. It drifts. It fractures. It fixes.
What makes the February corporate deck interesting is not the percentages. It is the pattern. A high-potential group was isolated: “Last-line ICI or ≤2 prior systemic treatments.” On its face, that reads like standard development housekeeping. In the Layer 2 frame, it reads differently. It reads as an architectural filter.
Line of therapy is not merely a clinical variable. It is a proxy for distance from the epigenetic boundary. Each successive systemic exposure increases the probability of nodal corrosion: dendritic relay inefficiency, stromal distortion, TCF1⁺ attrition, chromatin commitment. By the time a patient has traversed multiple lines, the node may no longer be a governance organ—it may be a scar. The February segmentation implicitly acknowledges that renewal requires residual structure. It does not say this explicitly. But the stratification echoes it.
Look at the monotherapy waterfall. If MDNA11 were simply an amplifier—another execution-layer intensifier—monotherapy would be expected to whisper while the combination shouted. That is not what appears. The monotherapy arm in this selected population shows meaningful tumor regression. That is precisely what the Layer 2 model predicts if substrate renewal is real. A renewal engine should generate signal before checkpoint multiplication is applied. It should not depend entirely on PD-1 blockade to express activity. In a structurally permissive node, restoration alone should widen the durability window. The data pattern is directionally concordant with that claim.
Then examine the combination. MDNA11 plus pembrolizumab shows deeper responses within that same architectural window. That is not redundancy. It is sequencing. Restoration first. Amplification second. Layer 2 precedes Layer 4. If progenitor pools are expanded and DNAM-1 competence is preserved, checkpoint blockade becomes a multiplier rather than a desperate rescue maneuver. The combo looks stronger because execution is now operating on substrate. That is consistent with an architectural sequencing model rather than accidental synergy.
Even the choice of expansion cohorts—MSI-H, TMB-H, melanoma, endometrial—aligns with the structural prerequisites described in the Layer 2 blow-out. These are not uniformly fibrotic, transport-collapsed deserts. They are contexts in which antigen traffic and organized immune microstructure often persist, at least partially. MDNA11 does not dissolve collagen. It does not regenerate HEVs. It does not rebuild FRC conduits. It requires permissivity. The indications selected implicitly reflect that constraint.
The important shift here is conceptual. The February deck provides an early clinical echo of the mechanistic thesis. It does not prove the governance model. It rhymes with it. Line-of-therapy becomes a crude but practical surrogate for architectural integrity. Earlier exposure correlates with a higher probability of remaining above the epigenetic fixation threshold. Monotherapy activity becomes a readout of renewal rather than mere effector intensification. Combination depth becomes evidence of proper layer sequencing rather than accidental synergy.
This is the difference between amplitude and architecture. Tumor shrinkage alone does not reveal which layer was engaged. But when monotherapy produces signal in a partially treatment-experienced population—and when that signal amplifies coherently with PD-1 blockade—it suggests that the intervention plane is upstream of execution. It is directionally consistent with substrate conditioning rather than pure terminal effector surge.
The IO Stack Layer 2 document argued that many immunotherapy disappointments represent transport or architectural failure misread as immune incompetence. The February segmentation quietly operationalizes that insight. By narrowing to ≤2 prior systemic treatments, the development program is, in effect, selecting for nodes more likely to retain dendritic relay and progenitor renewal capacity. It is not biomarker-level stratification. It is architectural staging by proxy. And that staging matters.
If durability depends on regenerative capacity, not peak activation magnitude, then early clinical signal should cluster where governance remains intact. If the epigenetic boundary has been crossed, no amount of amplifier pressure will restore renewal. The waterfall becomes a topographic map: some patients remain in green or yellow zones—structurally permissive with partial substrate depletion. Others have drifted toward orange or red—transport compromised or fixation complete. The responses segregate accordingly.
The deeper synthesis is this: the February deck does not require revision of the Layer 2 framework. It is directionally consistent with it. It provides the first faint clinical contour lines that align with the architectural cross-section. It suggests that restoration may behave differently than pure amplification in human tissue.
This is not proof of universal rescue. The blow-out made clear that MDNA11 cannot reverse epigenetic lock. It cannot rebuild destroyed nodes. Its domain is conditional restoration within a still-recognizable governance organ. The February segmentation implicitly accepts that boundary. It narrows the aperture to where restoration remains biologically plausible.
From architecture to waterfall. The model predicted that substrate restoration would manifest as early monotherapy signal in structurally permissive settings and that amplification layered on top would deepen response. The deck is broadly consistent with that pattern. Not conclusively. Not yet telemetrically validated with longitudinal nodal sampling or TCF1⁺ kinetics. But directionally coherent.
Layer 2 was never about explaining why everything works. It was about explaining why many things fail. The February data do not overturn that thesis. They begin to draw its clinical shadow. And shadows, when they align with structure, are rarely accidental.
PRISM-11 Translating Data To Architecture
This waterfall plot is typically read as magnitude — percent shrinkage from baseline. But viewed through the Layer 2 governance lens, it also encodes something else: architectural state. Earlier-line exposure and limited prior systemic therapy function as coarse proxies for residual lymph node integrity — for how far a patient may be from the epigenetic fixation boundary described in the Layer 2 blow-out. The clustering of deeper regressions in patients treated earlier in their therapeutic trajectory is directionally consistent with a renewal-first model rather than a pure amplifier effect.
MDNA11 monotherapy showing objective regressions in this population suggests substrate restoration is possible before execution-layer multiplication is applied. When checkpoint blockade is layered on top in less structurally compromised settings, response depth appears to increase — a pattern that aligns with sequencing discipline (Layer 2 before Layer 4), not redundancy.
This graphic does not prove TCF1⁺ preservation, dendritic relay restoration, or chromatin reversibility. It does something more modest but important: it shows a clinical pattern that coheres with a governance model in which durability depends on regenerative capacity, not peak activation alone. Architecture first. Amplification second.
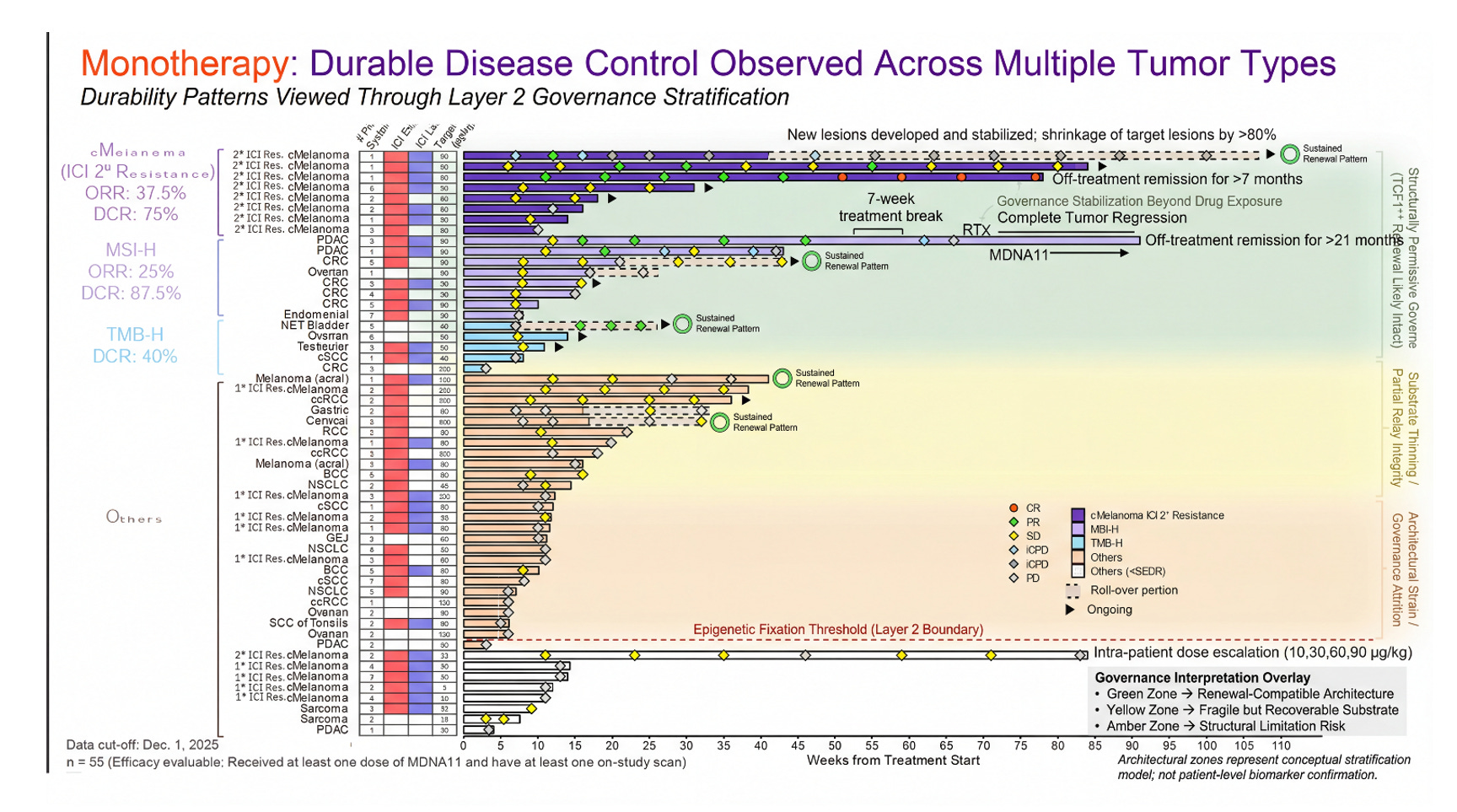
Figure: Objective Tumor Regression Viewed Through a Layer 2 Governance Lens
This waterfall plot presents best percent change in target lesions for MDNA11 monotherapy across tumor types, overlaid with a conceptual Layer 2 architectural stratification model. The vertical gradient does not reflect measured nodal biomarkers, but rather a governance hypothesis: deeper and more sustained regressions cluster within a renewal-compatible architectural window (green), whereas shallow responses or progression increasingly align with substrate thinning or structural attrition risk (yellow to amber). The dashed red line marks a conceptual “epigenetic fixation threshold,” below which residual progenitor integrity may be insufficient to sustain durable immune assembly without upstream restoration.
Importantly, this model reframes response heterogeneity as potentially architectural rather than purely pharmacologic. Apparent tumor shrinkage is visible at the execution layer; durability depends on whether tumor-draining lymph node governance remains permissive for TCF1⁺ renewal and rhythmic STAT5 signaling. The zones shown represent a stratification hypothesis — not patient-level biomarker confirmation — intended to illustrate how depth of regression and line-of-therapy context may intersect with upstream substrate integrity.
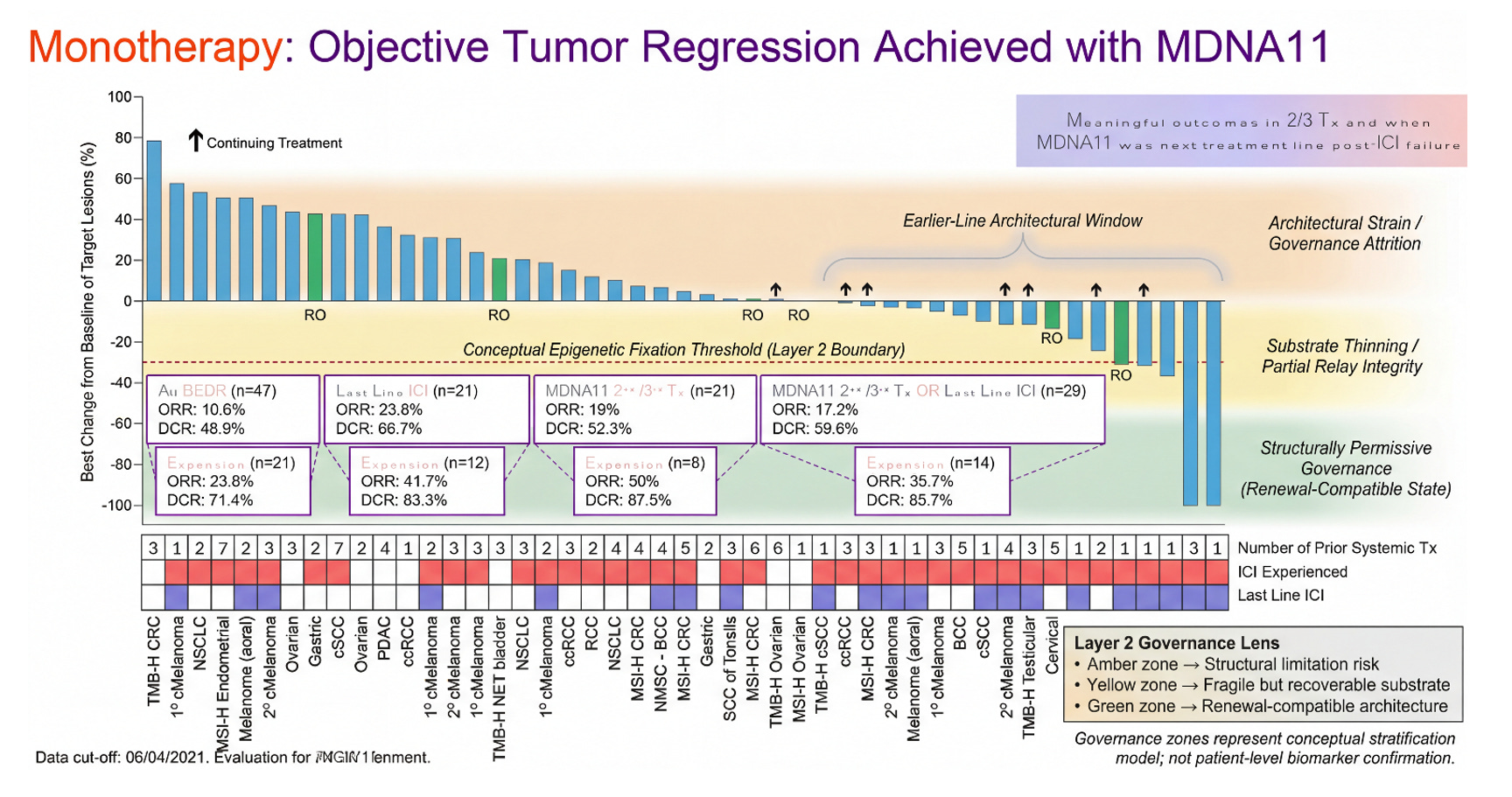
Monotherapy is where the architectural question becomes visible. In the Layer 2 framework, durability is not a function of peak activation magnitude; it is a function of renewal substrate integrity. The tumor-draining lymph node is not merely a relay station but a governance organ. Within it, stromal topology, chemokine zoning, TCF1⁺ progenitor preservation, and rhythmic STAT5 signaling determine whether effector deployment can be sustained or whether it collapses into transient flare.
MDNA11 monotherapy, viewed through this sub-layer logic, is not an amplifier. It is a substrate intervention. Its βγ selectivity, α-avoidance, and lymph node–resident distribution pattern align with progenitor stabilization rather than terminal differentiation pressure. When tumor regression occurs under monotherapy, the implication is not simply that effector cells were activated. It suggests that the renewal pool was sufficiently preserved to translate signal into sustained deployment. When regression is shallow or absent, the question becomes architectural: how much of the governance scaffold remains intact, and is it above the renewal threshold? This is the hinge.
Checkpoint blockade, by contrast, is not a builder. It is a conditional amplifier. PD-1 inhibition does not create progenitors; it releases inhibitory constraint on cells that already exist. Its performance therefore depends on the state of the upstream substrate. In a collapsed nodal environment — stromal distortion, TCF1⁺ depletion, epigenetic fixation — checkpoint blockade can appear mechanistically ineffective when in reality it is contextually stranded.
The rollover cohort becomes a live sequencing experiment. Patients first receive MDNA11 monotherapy — substrate reconditioning. Pembrolizumab is then introduced — execution-layer engagement. The key question is not whether responses occur, but whether depth or trajectory changes after amplifier addition. If regression deepens following PD-1 introduction in patients previously exposed to checkpoint inhibitors, one interpretation is conditional re-sensitization rather than simple additive cytotoxicity. It is conditional re-sensitization — amplifier function restored in the presence of renewed substrate integrity. This is not proof of mechanism. It is architectural coherence. The sequence matters because the layers matter. Substrate first. Amplifier second.
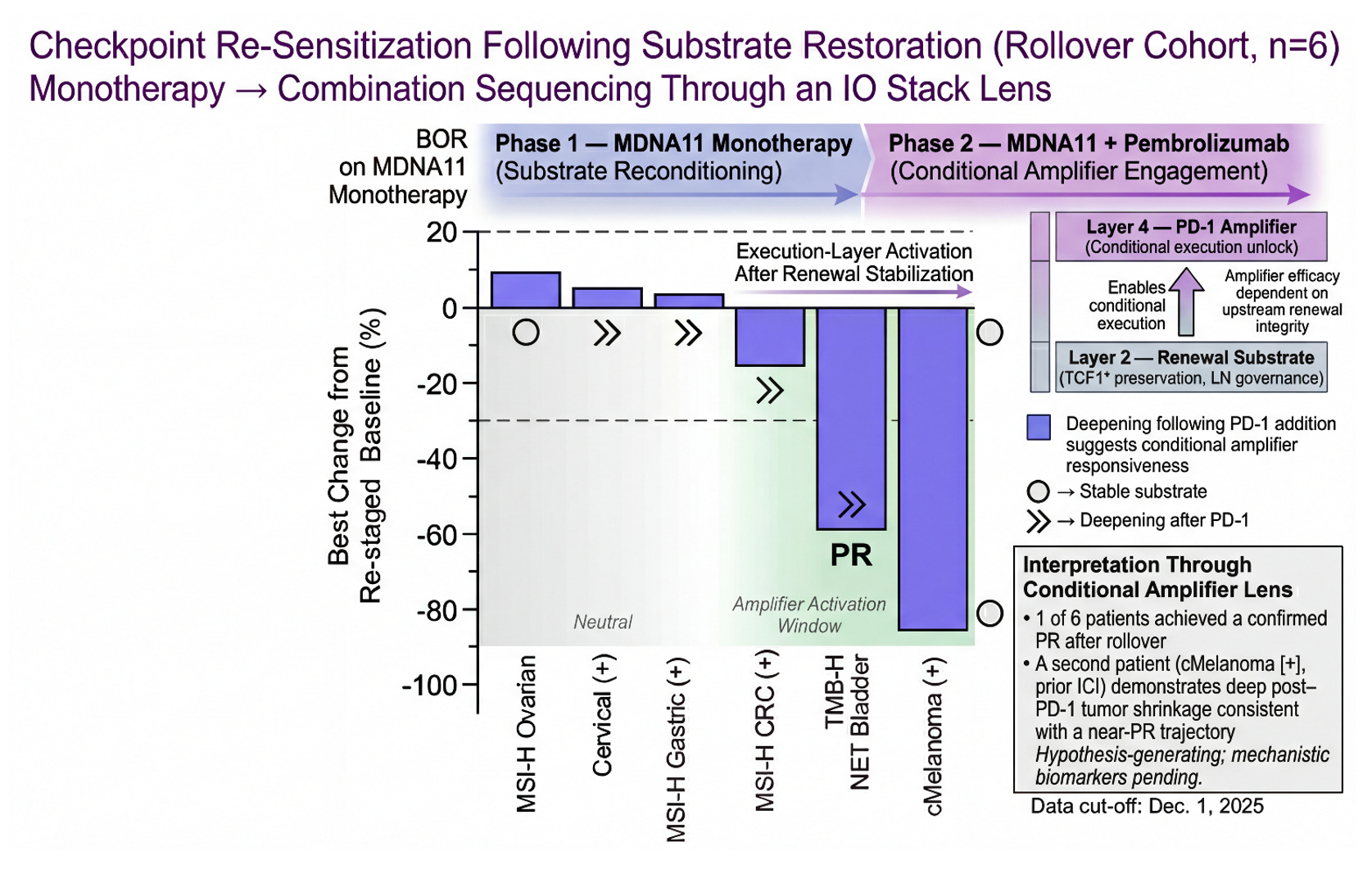
Checkpoint Re-Sensitization Following Substrate Restoration (Rollover Cohort, n=6)
This figure visualizes rollover patients through a Layer 2 → Layer 4 sequencing lens. Phase 1 (MDNA11 monotherapy) is framed as substrate reconditioning within the renewal architecture of the tumor-draining lymph node. Phase 2 (MDNA11 + pembrolizumab) represents conditional amplifier engagement at the execution layer. One patient achieved a confirmed PR following rollover, and a second cMelanoma (+) case demonstrates deep post–PD-1 shrinkage consistent with a near-PR trajectory. The pattern raises the possibility that checkpoint efficacy may be partially substrate-dependent rather than permanently lost after prior ICI exposure. The shaded “Amplifier Activation Window” illustrates where execution-layer release may translate into deeper regression when renewal integrity is preserved. These observations are hypothesis-generating and require mechanistic validation, but they align with a substrate-first model of immunotherapy sequencing.
The rollover cases tell a within-patient story. They show that something changes after exposure to MDNA11 monotherapy—stability becomes regression, shallow shrinkage becomes deeper contraction, and in at least one case a confirmed partial response emerges only after PD-1 is reintroduced. But case sequencing alone does not answer a broader question: is this an isolated phenomenon, or does the geometry of response shift at the cohort level once the amplifier is engaged? To address that, we have to zoom out from individual trajectories and look at distribution shape.
When the monotherapy waterfall is viewed on its own, it already demonstrates substrate activity. There is stabilization above the zero line, and there is a meaningful tail of regression below it. But the deep-response region is narrow. The execution layer appears intermittently active, not broadly engaged. Once pembrolizumab is layered in, however, the distribution changes character. The deep-regression tail widens. The lower end of the curve thickens. The geometry suggests that the execution layer is no longer sporadically firing—it is being conditionally unlocked.
The monotherapy dataset is where you can see the mechanism’s “signature” without help: MDNA11 is not just pushing transient effector expansion, it’s plausibly conditioning the renewal substrate that those effectors depend on. In IO-stack terms, that means the meaningful action is upstream of PD-1 execution—Layer 2 governance and its sublayers (TDLN anchoring, progenitor preservation, rhythmic STAT5 reinforcement, relay integrity). When Layer 2 is intact, downstream amplifiers look powerful; when Layer 2 is corrupted or exhausted, amplifiers become stranded—capable of releasing brakes, but with too little organized substrate left to translate that release into durable control. This is why “depth” and “durability” in monotherapy matter: they’re not just outcomes, they’re proxies for whether the architecture is being restored versus merely heated up. Once you accept that framing, the combination data stops being a generic “combo works better” story and becomes something sharper: PD-1 doesn’t create the substrate—PD-1 exploits it. The right way to read MDNA11→pembro sequencing is therefore not as additive cytotoxicity, but as a conditional execution unlock that becomes visible only after the renewal layer has been stabilized. That is the logic behind the next slide: we’re not claiming a formal cross-cohort statistical comparison; we’re showing an architecture-consistent pattern where the deep-response tail expands when a conditional amplifier is engaged on top of a conditioned substrate—especially notable when that expansion includes rollover patients with prior ICI exposure.
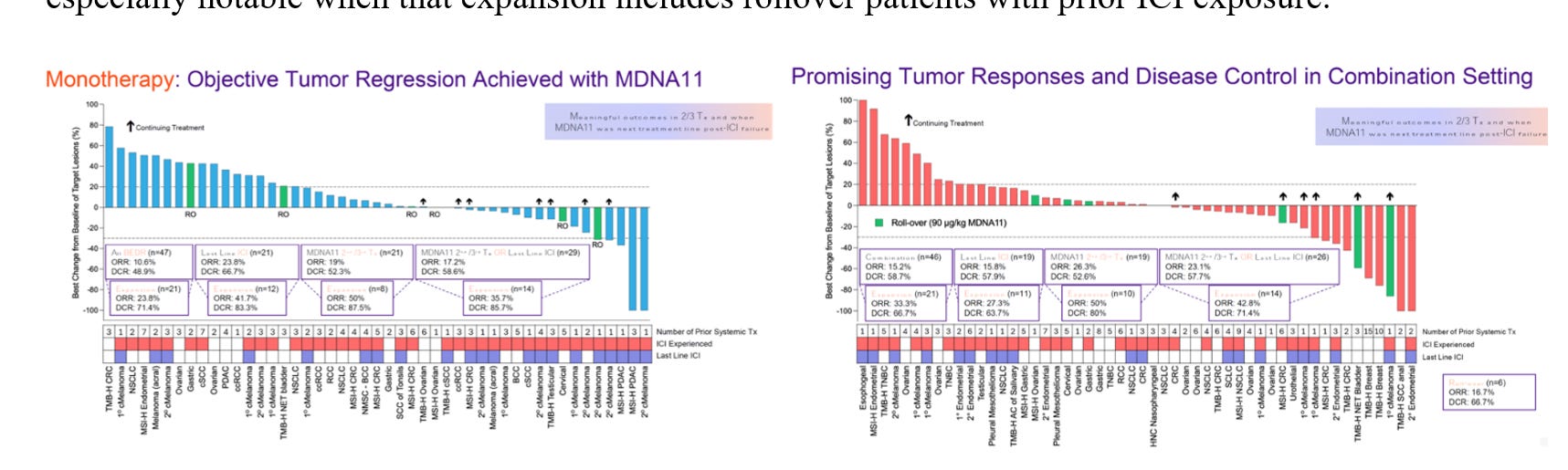
Crucially, this expansion is not confined to checkpoint-naïve biology. A portion of the deepening responses occurs in rollover patients, four of whom had prior ICI exposure. That does not prove re-sensitization mechanistically, but it is consistent with a model in which PD-1 function depends on upstream renewal integrity. In this framing, MDNA11 is not competing with the checkpoint; it is conditioning the architecture that allows the checkpoint to execute. The slide that follows is therefore not a statistical comparison between cohorts. It is a structural one: a visualization of how depth-of-response geometry shifts when substrate conditioning precedes conditional amplifier engagement.
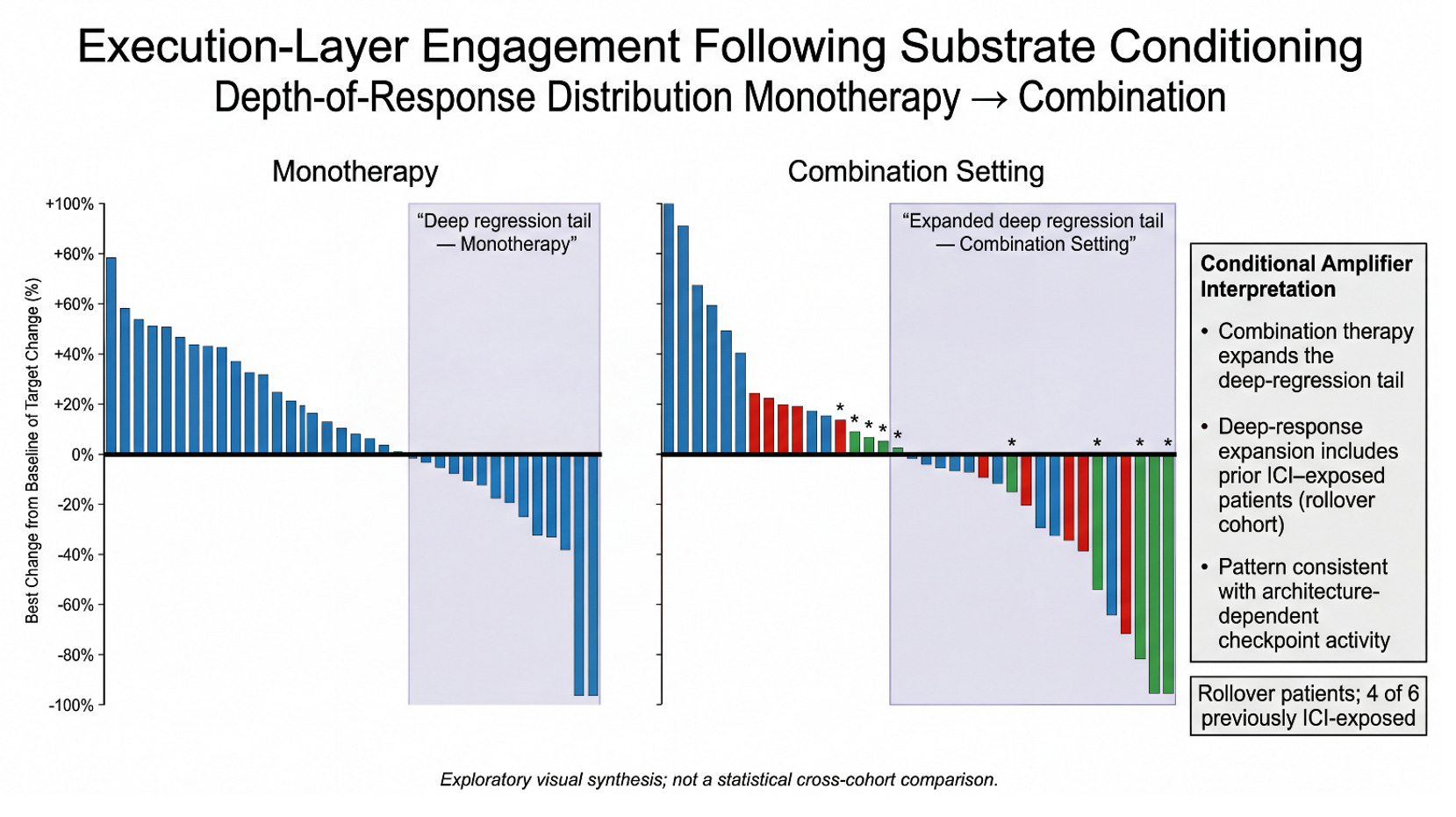
This figure visualizes a simple architectural hypothesis: MDNA11 conditions the renewal substrate, and pembrolizumab acts as a conditional amplifier whose execution becomes meaningful only once that substrate is sufficiently stabilized. In monotherapy (left), the response distribution shows a visible “deep regression tail,” consistent with MDNA11’s intrinsic ability to drive tumor shrinkage in a subset without checkpoint assistance. In the combination setting (right), the deep-response tail appears expanded, suggesting that PD-1 engagement is converting substrate conditioning into more frequent and/or deeper execution-layer outcomes. Importantly, the expanded deep tail includes rollover patients, and 4 of 6 rollover patients were previously ICI-exposed—supporting a re-sensitization read rather than a naïve-only effect. The interpretive claim is not that the cohorts are statistically matched, but that the pattern aligns with an IO-stack model where PD-1 efficacy is architecture-dependent. Put differently: the combo plot looks like what you’d expect if MDNA11 is restoring the “governance layer” that conditional amplifiers require, rather than simply adding another independent mechanism of killing.
The depth-of-response distribution shows the pattern. The matrix asks whether that pattern is structured. In the prior slide, we observed that combination therapy appears to expand the deep-regression tail relative to monotherapy — and that this expansion includes rollover patients, many of whom were previously ICI-exposed. But distribution alone does not tell us whether this is random scatter or architecture-dependent behavior. To interrogate that question, we reframe the cohort through a two-dimensional lens: substrate integrity as proxied by line-of-therapy stress, and amplifier engagement depth as measured by best percent change. If PD-1 is behaving as a conditional amplifier — rather than an independent driver — we should expect deep execution to cluster where substrate integrity is preserved, but not be strictly confined to it. The 3×3 matrix below does not change the underlying data. It reorganizes it to test whether execution-layer depth tracks with architectural stress gradients — and whether rollover patients disproportionately occupy the deep band. What emerges is not a claim of statistical proof, but a structural signal.
This 3×3 matrix reorganizes the full combination cohort (n=46) along two biologically motivated axes: substrate integrity (line-of-therapy stress) and amplifier engagement depth (best percent change). The horizontal axis stratifies patients by prior systemic therapy exposure (≤2, 3–4, ≥5), serving as a pragmatic proxy for immune architectural attrition. The vertical axis captures depth of execution, distinguishing deep regression (≥50%), intermediate regression (20–49%), and non-deep responses, which include stable disease and modest shrinkage. Deep responses are distributed across all substrate stress states, including heavily pretreated patients (≥5 prior therapies), indicating that execution-layer activity is not confined to earlier-line disease. Notably, rollover patients are disproportionately represented in the deep band (2 of 3), and 4 of 6 rollover patients were previously ICI-exposed — consistent with a re-sensitization framing rather than naïve-only activity. Importantly, only 9 of 46 patients exhibited >20% tumor growth, while 37 of 46 demonstrated ≤20% growth or tumor shrinkage, contributing to an overall disease control rate of 58.7% in the combination cohort. The matrix can be read as reframing the majority “non-deep” band not as absence of activity, but as heterogeneous biological responses beneath the deep-regression threshold. While exploratory and non-statistical, the pattern is directionally consistent with an architecture-dependent, conditional amplifier model of PD-1 engagement following substrate conditioning.
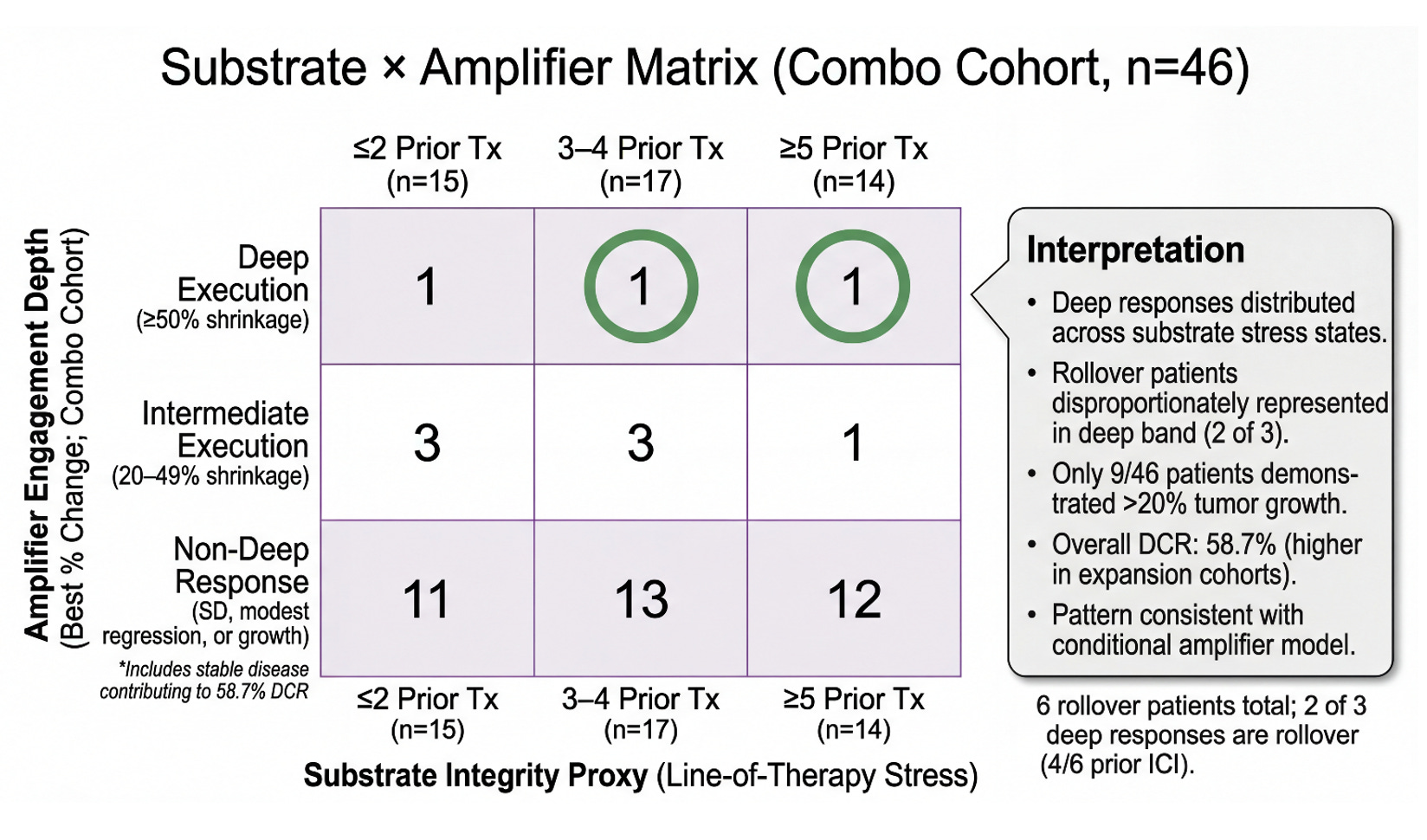
Contextual Notes (Combo Cohort, n=46)
· 23/46 patients showed tumor shrinkage
· 9/46 patients (20%) had >20% tumor growth.
· 37/46 patients (80%) had ≤20% growth or tumor shrinkage.
· Overall DCR: 58.7%.
· Expansion cohorts demonstrated DCR up to 80%.
· Deep regression observed across all line-of-therapy strata.
· 2 of 3 deep responses occurred in rollover patients (4/6 prior ICI).
Case Signal: When 10–15 Lines of Therapy Do Not Equal Immune Collapse
Buried inside the combination waterfall and swimmer plots are two patients who should not exist under a naïve “lines-of-therapy = immune exhaustion” model. Both are TMB-H breast cancer (non-TNBC). Both are ICI-naïve. And both carry extraordinary prior systemic treatment burdens — one with 10 prior lines, the other with 15. Under conventional oncology heuristics, that degree of treatment exposure typically implies biologic aggressiveness, clonal heterogeneity, cumulative marrow suppression, and immune disorganization. Yet in the combination cohort, both patients achieved deep tumor shrinkage in the ~65–80% range.
If prior line count were a direct surrogate for immune architectural collapse, these bars should not appear on the far right tail of the waterfall. But they do. This is where the Layer 2 sublayer model becomes clinically explanatory rather than theoretical. Line count is not equivalent to governance failure. Checkpoint exposure is not equivalent to chemotherapy exposure.
A patient can accumulate cytotoxic, endocrine, targeted, or antibody therapies and still preserve aspects of lymph node governance architecture — particularly TCF1⁺ progenitor pools and dendritic relay integrity — if PD-1–mediated exhaustion cycling has not already been chronically imposed. In contrast, repeated checkpoint cycling in the absence of substrate restoration risks converting partial impairment into fixation.
These two breast cancer patients represent extreme line-of-therapy stress without prior checkpoint amplifier cycling. Their immune systems were battered, but not amplifier-scarred. And when MDNA11 conditioned the substrate — followed by PD-1 engagement — the execution layer behaved like an amplifier again. This is not a “breast cancer story.” It is a stress test of the conditional amplifier thesis under maximal clinical burden.
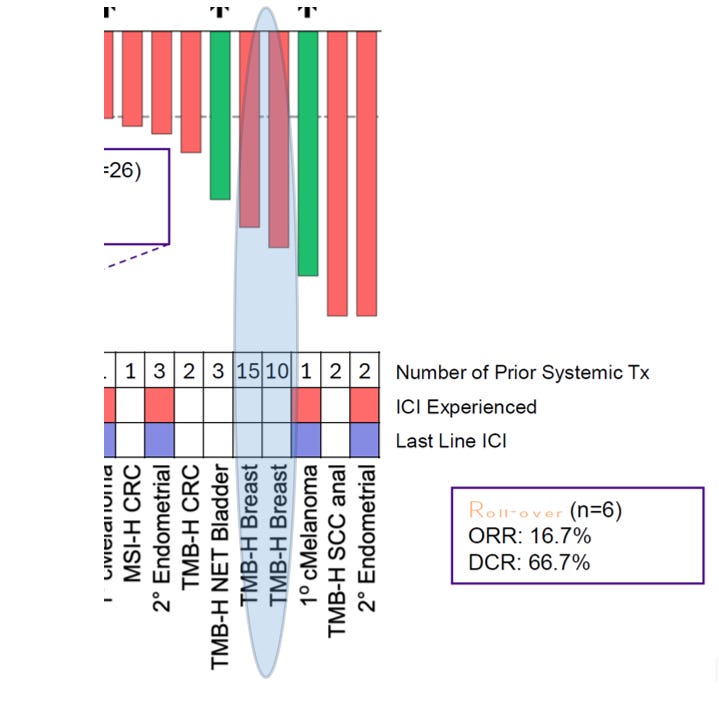
Layer 2 Governance Under Extreme Prior Therapy Load
The Layer 2 governance diagram at the beginning of this article was not decorative. It was structural. MDNA11 does not repair endothelial gatekeeping. It does not reverse stromal fibrosis. It does not undo epigenetic terminal fixation once chromatin locks are complete. Its operative zone lives in the renewal corridor — dendritic relay support, TCF1⁺ progenitor stabilization, rhythmic STAT5 reinforcement.
The two TMB-H breast patients appear to sit in an intermediate Layer 2 state:
• Structural throughput intact
• No irreversible fixation
• Progenitor pool reduced, but not extinguished
• No prior checkpoint-driven exhaustion cycling
That matters. Because in this configuration, a β-selective IL-2 signal can expand and stabilize progenitors upstream of PD-1 engagement. Once the amplifier is introduced, it is pulling on renewable substrate rather than an exhausted terminal population. The magnitude of shrinkage in these two patients is therefore not simply a “deep response.” “It suggests that Layer 2 governance may persist despite extraordinary systemic therapy exposure— provided the epigenetic fixation threshold has not been crossed.
This aligns directionally with the substrate × amplifier matrix we just constructed. Deep execution is not restricted to ≤2 prior lines. It appears across substrate stress states when structural permissivity remains. And critically — both of these patients were ICI-naïve. That distinction is not cosmetic.
Why This Reinforces the Salvage Valuation Thesis
Ten months ago, in A Billion-Dollar Layup in Salvage Therapy, the valuation floor was built on a conservative premise: that MDNA11 did not need to win first-line dominance to justify material enterprise value. It needed to carve out defensible salvage wedges in biomarker-defined, high-unmet-need populations where substrate restoration could convert checkpoint failure into response.
What these two patients suggest is that the salvage wedge may be broader than originally modeled. Salvage is not only “post-ICI failure.” Salvage also includes “post-everything-else, pre-checkpoint.” In other words, late-line does not always mean checkpoint-refractory. There exists a subset of chronically treated patients who have exhausted cytotoxic and targeted options but have not yet biologically burned through PD-1 as an amplifier. For them, the MDNA11 + PD-1 pairing is not rescue — it is first meaningful execution.
From a commercial standpoint, that enlarges the salvage surface area. From a regulatory standpoint, it supports the argument that line count alone should not be used as a negative substrate proxy. From a mechanistic standpoint, it validates the Layer 2 governance thesis under extreme clinical stress. And from a valuation standpoint, it strengthens — not weakens — the original conservative $1B salvage framing. Because if deep responses can occur in 10- and 15-line patients without prior checkpoint exposure, the total addressable salvage architecture is not limited to CPI-refractory niches. It includes CPI-naïve late-line populations stratified by immunogenic biomarkers such as TMB-H. That is a larger wedge than originally modeled.
Important Boundary Conditions
This is exploratory signal, not randomized proof. n = 2 is not a registrational dataset. We do not have full molecular granularity beyond TMB-H labeling on the slide. But in mechanistic modeling, anomalies matter. Especially when they violate conventional heuristics. If 15 prior lines do not preclude 70% tumor shrinkage once substrate is conditioned and PD-1 engaged, then the narrative that “late line equals immunologic futility” is overly simplistic. And if that narrative is overly simplistic, the valuation discount historically applied to late-line cytokine programs may also be overly simplistic. That is the deeper implication.
The Salvage Denominator Was Probably Too Small
Ten months ago, in A Billion-Dollar Layup in Salvage Therapy, the valuation floor for MDNA11 was built deliberately conservatively. The structure was simple and intentionally restrained. We limited the salvage addressable population in North America and Europe to three buckets: MSI-H/dMMR (~50k), CPI-resistant (~85k), and cold tumors (~20k). Then we cut that aggregate in half to avoid optimism creep. That produced ~155,000 salvage-eligible patients. At $100,000 per patient per year, that implied a $15.5B salvage TAM. At 10% penetration, peak revenue was modeled at ~$1.55B. Discounted under a cautious ramp and 10% cost of capital, salvage alone supported an NPV of approximately $6.3B.
The key feature of that model was not aggressiveness. It was discipline. But there was a quiet structural assumption embedded inside it: that “salvage” was functionally synonymous with CPI-experienced or CPI-refractory biology. The two TMB-H breast cancer patients in the combo waterfall complicate that assumption.
Both had 10 and 15 prior systemic lines. Both were ICI-naïve. Both demonstrated deep tumor shrinkage in the 65–80% range. Under conventional heuristics, a patient with fifteen prior regimens is clinically salvage by any rational definition. Yet biologically, these patients were not CPI-refractory because they had never received CPI. They were late-line, but not amplifier-burned. That distinction matters economically.
If salvage is defined by line-of-therapy stress and unmet need — not merely by checkpoint history — then the denominator in the original model was likely under-inclusive. There exists a subset of CPI-naïve, heavily pretreated, biomarker-selected patients (for example TMB-H) who are clinically salvage but were not explicitly counted in the 2025 framework. The clean way to test this is not to inflate assumptions, but to run a sensitivity on the existing scaffold.
Base case (from Billion-Dollar Layup):
Salvage-eligible patients (post-haircut): ~155,000
TAM at $100k/pt-year: $15.5B
Peak revenue at 10% penetration: $1.55B
Salvage NPV proxy: ~$6.3B
Now ask one question: what if CPI-naïve, late-line biomarker populations add only a modest increment to that denominator? If the inclusion of CPI-naïve late-line patients expands the salvage pool by just:
• 5% → TAM rises to ~$16.3B → peak revenue ~$1.63B → salvage NPV ~$6.6B
• 10% → TAM ~$17.1B → peak revenue ~$1.71B → salvage NPV ~$7.0B
• 20% → TAM ~$18.6B → peak revenue ~$1.86B → salvage NPV ~$7.6B
• 30% → TAM ~$20.1B → peak revenue ~$2.0B → salvage NPV ~$8.2B
Nothing else changes. Price unchanged. Penetration unchanged. Ramp unchanged. Discount unchanged. This is not a new TAM claim. It is a denominator sensitivity layered onto the exact conservative architecture built ten months ago.
And what makes the two TMB-H breast patients relevant is not the tumor type per se. It is that they demonstrate a clinically salvage state (10–15 prior lines) without prior checkpoint cycling. In Layer 2 governance terms, they represent extreme systemic stress without confirmed epigenetic fixation. When MDNA11 conditions substrate and PD-1 is introduced for the first time, execution can still occur.
If that biology generalizes even modestly, the salvage wedge is broader than “post-CPI failure.” It includes “post-everything-else, pre-CPI exhaustion.” That is not a speculative stretch. It is a definitional clarification. Late-line does not always equal checkpoint-refractory. And if late-line CPI-naïve biomarker-selected patients belong inside the salvage frame, then the conservative $1B floor thesis becomes structurally stronger — not because we changed assumptions, but because we realized the original ones may have been too narrow. The Layup thesis was built to survive pessimism. This sensitivity shows it may also survive inclusion.
Durability does not live in peak activation. It lives in substrate integrity. MDNA11 is designed to rebuild the engine that determines whether the immune system can sustain war.
There Is a Deeper Implication Beyond Numbers
The deeper implication is not merely that the salvage denominator may have been too small. It is that the Q1 2026 combination pattern begins to hint at something more systemic. MDNA11 monotherapy already shows that substrate-level activity can manifest before checkpoint multiplication is applied. When pembrolizumab is layered on top, the response distribution appears to deepen and widen — including in rollover patients and prior-ICI settings — in a way that is more consistent with conditional amplifier engagement than with generic additive cytotoxicity. In that framing, the relevant question is no longer only how many late-line patients fit inside a salvage bucket. It is whether substrate conditioning is increasing the amount of immune architecture available for downstream execution.
If Layer 2 is the renewal engine, then the natural next metric is not peak shrinkage but extension capacity: how far restored immune production can propagate across a metastatic system before depletion, organ context, and routing failure interrupt it. That is the logic of the Node Extension Score.
A therapy that restores progenitor renewal does more than rescue isolated patients; it may increase the number of lesions, niches, and metastatic sites that the immune system can cover at once. The valuation consequence is therefore not confined to a larger salvage denominator. It may lie in a larger immune operating radius. Layer 3 picks up precisely there: not at the point of activation, but at the point where restored substrate must be converted into distributed tumor control across the metastatic network.
Disclosure Statement
The author is an investor in Medicenna Therapeutics and licenses the PRISM-11 analytical framework to Medicenna for internal and external use with attribution. The author is not an employee of Medicenna. All views expressed are independent and for informational purposes only.
Thank you David - throwing darts here, but does Medicenna’s push for MDNA113’s IND this year (in light of their fiscal constraints & other needs - pivotal trial for MDNA11, for example) perhaps underscore the idea that a combination approach for treatment is now the focus?
Apologies in advance if you’ve already highlighted this elsewhere - still working on a thorough understanding of your output.