
TIGIT and the Stranded Amplifier Problem
Why Substrate Repair Changes the Economics of Downstream Immuno-Oncology

Author’s Note:
Acknowledging upstream governance and lymph-node integrity as I wrote about here, here, and here, frames what the primary determinants are that reshape several aspects of immuno-oncology:
· Biomarkers are reordered, not replaced: Focus shifts to indicators of upstream assembly rather than solely tumor-centric markers.
· Early kinetics become interpretable rather than noisy: Initial responses can now be linked to foundational processes.
· Non-response becomes classifiable: Distinctions emerge between suppressed (potentially reversible) and destroyed architecture.
· Trial design shifts from delayed inference to mechanistic tracking: Emphasis on real-time monitoring of upstream layers.
· Competitive landscapes collapse into substrate creators vs. substrate consumers: Therapies are stratified by whether they build immune capacity or merely exploit it.
This is not an additive paradigm; it is the organizing principle that renders existing data (and deals) coherent from a PRISM-11 lense. What I did in the BMS / Janux article is use the deal as a diagnostic test of the industry’s current mental model.
A masked T-cell engager is an execution optimization: it improves where and how killing happens. That can be enormously valuable. But it sits downstream in the control stack — it presumes the immune system has enough competent, renewable effector capacity to deploy in the first place. In PRISM-11 terms, it’s a refinement of deployment and local permissioning, not a rebuild of the immune assembly line. So the point of mapping Janux into the IO Control Stack is not to dismiss it; it’s to clarify what category of problem it solves, and what category it does not.
That distinction matters because it’s the same category error that keeps repeating across immuno-oncology: we keep capitalizing conditional activators as if they can manufacture substrate. Most of the time, they can’t — they can only multiply what already exists. That’s the bridge into the next two pieces you’re about to read on TIGIT. This idea that “TIGIT wasn’t wrong” is really shorthand for “amplification wasn’t wrong; it was mis-sequenced.” And that, in turn, tees up the next iteration of the Operational Playbook: if the industry is going to keep funding exquisite deployment machinery, we need a repeatable way to measure whether the upstream assembly system is intact, repairing, or collapsing — and to know that in weeks, not quarters.
If you assume the immune substrate in a patient is intact, then amplifiers like PD-1, TIGIT, CTLA-4, etc. should behave like multipliers — and you’ll interpret failure as “the target didn’t work.” But if you assume the substrate is degraded — if renewal competence in the lymph-node/TLS axis has already drifted into terminal programs — then amplification failure is not evidence against the target. It’s evidence that the multiplicand is gone. TIGIT didn’t have to be “wrong” for the program-level bet to fail; it only had to be deployed into biology that no longer had the renewable machinery TIGIT depends on.
That’s why I keep returning to lymph-node integrity (courtesy of the Novartis research), routing coherence, and progenitor renewal as upstream governance variables. They don’t replace biomarkers; they reorder them. They don’t make early kinetics less noisy by magic; they make early kinetics interpretable because you can link the signal to a mechanistic lane. And they don’t just help you explain non-response — they let you classify it: suppressed and potentially reversible versus structurally destroyed.
The rest of this TIGIT 2-part discussion is essentially a sequencing argument inside that control stack. The 2nd part (which will post Friday) discusses the capital allocation and valuation implications. These will be followed by, “The Operational Playbook” revision article that refreshes and revises the practical layer: what do we measure, when do we measure it, and how do we update our confidence in real time instead of waiting 12 weeks to learn whether we were betting into plasticity or into fate-locked resistance.
Reclassifying the TIGIT “Failure”: Misplacement, Not Invalid Biology
The story of TIGIT is often told as a disappointment: a promising checkpoint axis that failed to live up to early expectations, consuming roughly $2.4 billion in capital before collapsing under a wave of negative Phase 2 and Phase 3 readouts. In this telling, TIGIT is grouped with a long list of “next PD-1s” that simply did not work.
That framing is convenient — and wrong. TIGIT did not fail because the biology was incorrect.
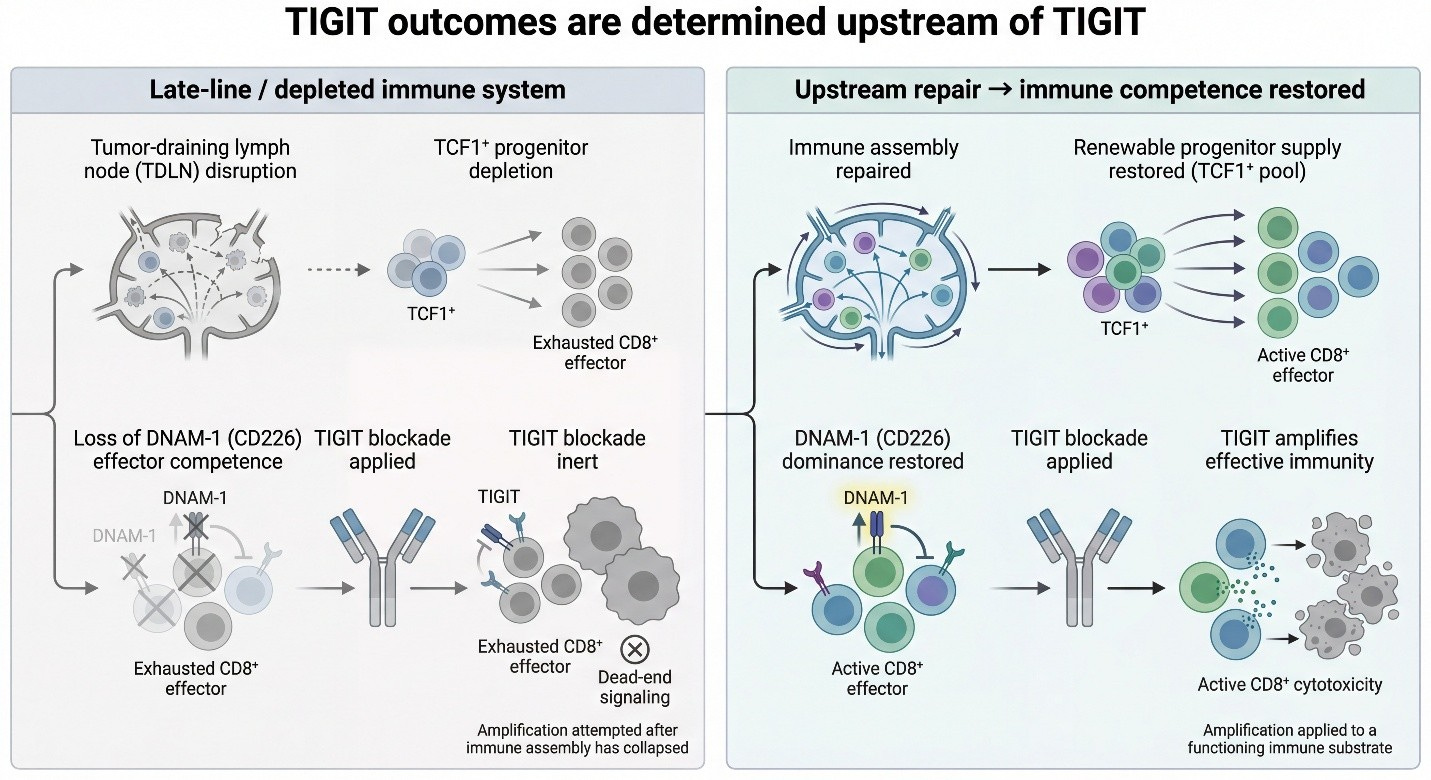
TIGIT failed because it was deployed too late, into systems where immune assembly had already collapsed. TIGIT blockade is only meaningful when CD226 (DNAM-1) signaling is still available to carry the activating signal once TIGIT is removed.
This distinction matters, because it separates mechanistic invalidation from contextual misplacement. The former kills an axis permanently. The latter implies that the axis may still function — but only when the structural prerequisites for its activity are intact.
At a mechanistic level, TIGIT has always made a narrow and specific assumption about the immune system it is acting upon. It does not create immune activity. It does not assemble effector populations. It does not restore immune memory. TIGIT is a regulatory brake operating at the tumor interface — a final-mile control point that modulates how existing effector cells behave once they arrive.
Implicit in that role are three non-negotiable assumptions:
DNAM-1⁺ effector cells must already exist
TIGIT antagonizes DNAM-1 (CD226) signaling through shared ligands such as CD155 (PVR) and CD112. If DNAM-1–competent CD8⁺ T cells or NK cells are absent, TIGIT blockade has nothing to “release.” There is no latent cytotoxicity to unmask.Those effectors must be renewable, not terminal
TIGIT signaling predominates in chronically stimulated, exhausted environments. If the effector pool is terminally differentiated — disconnected from TCF1⁺ progenitor supply — then transient relief of inhibition cannot translate into durable tumor control. The system consumes what little immune capital remains and then collapses again.Those effectors must be able to reach the tumor
TIGIT acts at the immune synapse. If lymphatic routing, endothelial access, or stromal architecture prevent coherent infiltration, then TIGIT blockade operates upstream of nothing. Signal is removed, but no cells arrive to act on it.
A practical diagnostic rule follows from this. A TIGIT result is only interpretable if the system still satisfies three upstream conditions: (1) DNAM-1⁺ effector competence exists, (2) renewable progenitor supply remains (i.e., the TCF1⁺ engine is not depleted), and (3) trafficking from tumor-draining lymph nodes to tumor is intact. If any one of these is broken, failure does not falsify TIGIT biology—it only confirms that amplification was attempted after assembly had already collapsed. In that sense, many late-line TIGIT readouts were not “negative trials.” They were structurally non-diagnostic experiments. When viewed through this lens, the outcome of late-line TIGIT trials becomes almost inevitable.
Most TIGIT programs were deployed after PD-1 failure, often in heavily pretreated patients whose tumor-draining lymph nodes had been functionally exhausted, surgically disrupted, or immunologically silenced. In these settings, DNAM-1⁺ effector populations were already depleted, renewal pathways were broken, and trafficking was inconsistent at best. TIGIT blockade was asked to perform immune resuscitation — a task it was never designed to do.
The result was predictable: no signal, no durability, no survival benefit. This does not indict the TIGIT axis. It indicts the assumption that all checkpoints behave like PD-1, capable of generating activity de novo in depleted systems. TIGIT is not PD-1.
Reframed this way, the $2.4B “failure” of TIGIT is no longer a referendum on the biology of the nectin axis. It is a case study in chronological error — an intervention applied after the immune substrate it depends on had already eroded. TIGIT didn’t fail because it was weak; it failed because it was asked to amplify a signal whose source had already been extinguished.
This reclassification opens the door to a more important question, and one that the original TIGIT programs never asked: what would TIGIT have looked like if it had been deployed into a repaired immune system — one with renewable, DNAM-1–competent effector supply and intact upstream assembly? With that distinction in place, the question shifts from “Was TIGIT real?” to “What did TIGIT require in order to be real in patients? That question is where the story actually begins.
This is the structural mistake that defined the TIGIT era. TIGIT blockade was repeatedly deployed as if it were a generator of immune activity, when in fact it is only an amplifier of immune state. In systems where tumor-draining lymph nodes are disrupted, progenitor renewal has collapsed, and DNAM-1–competent effector populations no longer exist, TIGIT has nothing left to act on. Under those conditions, blockade is mechanistically intact but biologically inert.
By contrast, when immune assembly is repaired upstream—restoring TCF1⁺ progenitor supply and re-establishing DNAM-1 dominance—TIGIT functions exactly as designed: not by creating immunity, but by amplifying an already competent effector substrate. The figure below illustrates this bifurcation. TIGIT outcomes are determined before TIGIT is ever applied.
Why Downstream Checkpoint Extensions Failed to Scale: Amplification Without Assembly
The TIGIT experience is not an isolated case. It exemplifies a broader structural limitation that has constrained multiple checkpoint-adjacent strategies: downstream amplification presumes intact upstream immune architecture.
TIGIT blockade, PD-1–tethered cytokines, and tumor-conditional immune agonists all depend on viable, renewable effector T-cell populations already present in or near the tumor microenvironment. These approaches can enhance signaling intensity, but they cannot reconstruct immune capacity when lymph-node priming is deficient.
Clinical TIGIT data make this limitation visible. Robust target engagement consistently yielded inconsistent, non-durable, and context-dependent responses — exactly what would be expected when deployment-phase modulation is applied to patients with compromised tumor-draining lymph nodes due to prior therapy, disease burden, metabolic stress, or stromal disruption.
Similar constraints apply to bispecific antibodies and tethered cytokine platforms. While effective in immune-competent or early-line settings, these modalities rely on a functioning progenitor supply chain from the lymph node. When that supply chain is broken, downstream amplification does not scale, regardless of molecular sophistication.
The competitive landscape therefore stratifies not by molecular elegance, but by biological positioning. Some therapies assume immune competence; others attempt to restore it. TIGIT’s failure underscores that distinction. Durable efficacy cannot be achieved by amplifying what has not been structurally assembled.
Engineering Cannot Replace Biology
The TIGIT field implicitly believed that better antibody design could compensate for biological context. Fc silencing, affinity tuning, bispecific formats — all were pursued with the assumption that the right molecular tool could unlock latent immunity.
But TIGIT is not a generator of immune activity. It is a regulator of competition between inhibitory and activating receptors. Most re-sensitization studies focus on identifying alternative inhibitory receptors or microenvironmental modulators, without addressing the prerequisite of immune competence. This omission explains why many mechanistically sound interventions fail to generalize clinically.
No amount of antibody optimization can restore:
DNAM-1 competence once it is transcriptionally or post-translationally lost
Progenitor renewal once lymph node architecture has collapsed
Effector supply once immune capital has been consumed
This is why Fc-silent antibodies showed cleaner safety profiles and better pharmacology, yet failed to produce consistent clinical rescue. They addressed how TIGIT blockade was delivered, not whether the system was still capable of responding.
The Missing Assumption
Why PD-1 Expression Was the Wrong Enrichment Strategy
PD-1 expression is a marker of prior activation and chronic stimulation, not of functional killing capacity. In late-line tumors, high PD-1 often reflects a population of exhausted, terminally differentiated cells that are disconnected from progenitor renewal and stripped of activating receptor competence. In contrast, TIGIT blockade only has functional consequences if DNAM-1 signaling remains intact.
This is not a subtle mechanistic nuance — it is a hard requirement. TIGIT suppresses DNAM-1 through higher-affinity ligand engagement and inhibitory signaling. If DNAM-1 expression is lost, degraded, or uncoupled from downstream signaling, then TIGIT blockade removes an inhibitor from a pathway that is already nonfunctional.
In that context, PD-1 positivity becomes actively misleading. It selects for patients who appear immunologically “hot” by transcriptomics, yet lack the activating receptor machinery TIGIT depends on.
Here’s the diagnostic inversion TIGIT forced on us. PD-L1 asks whether the tumor is visible to the immune system. DNAM-1 asks whether the immune system is still capable. Late-line TIGIT trials often enriched for the former while unknowingly selecting against the latter. In other words: they chose patients whose tumors looked “immune-engaged,” while the immune engine required for TIGIT to matter—DNAM-1-competent effectors fed by renewable progenitors—had already been depleted.
DNAM-1 / CD226 Status Predicts TIGIT Response — Mechanistically, Not Retrospectively
Across multiple independent studies — spanning CD8⁺ T cells and NK cells, murine and human systems, and both tumor and peripheral compartments — a consistent pattern emerges:
TIGIT-expressing cells that retain DNAM-1 competence can recover cytotoxicity when TIGIT is blocked.
TIGIT-expressing cells that lack DNAM-1 signaling capacity do not respond, regardless of TIGIT expression level or ligand abundance.
Loss of DNAM-1 function — through transcriptional downregulation, proteasomal degradation, or chronic ligand engagement — is sufficient to render TIGIT blockade inert.
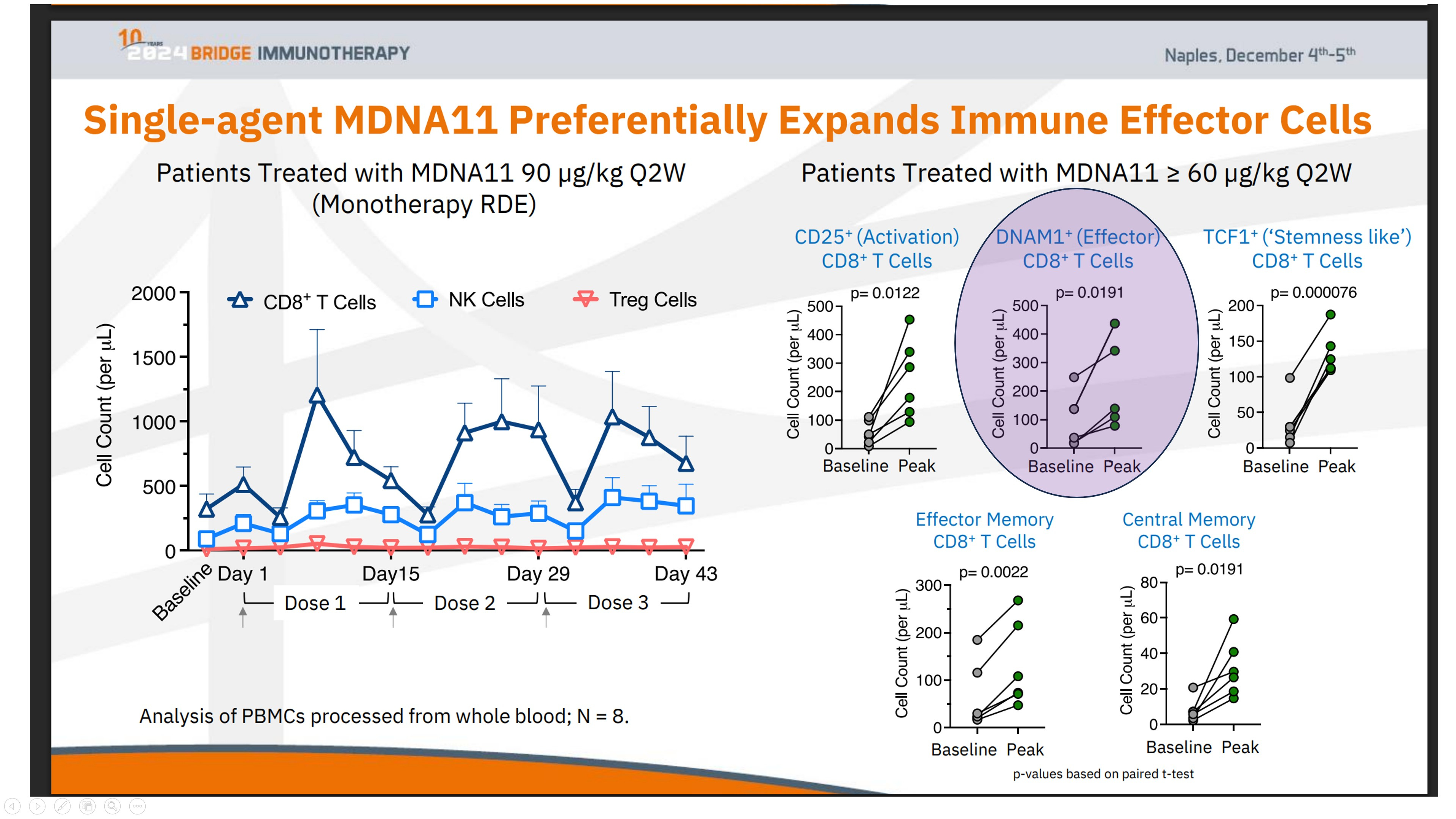
This explains a long-standing paradox in the TIGIT literature: why TIGIT blockade appears potent in early or less-treated models, yet collapses in advanced disease despite abundant ligand expression. The determinant is not whether TIGIT is present. It is whether DNAM-1 is still operational. In other words, DNAM-1 competence is not a correlative biomarker — it is the mechanistic switch that decides whether the nectin axis is actionable at all.
Why Ligand Density Without DNAM-1 Context Is Meaningless
Many TIGIT trials and translational analyses focused heavily on ligand expression: CD155 (PVR), CD112, and related nectins. The assumption was straightforward — high ligand density should predict benefit from TIGIT blockade. But ligand geography only defines which receptor dominates, not whether activation can occur.
High PVR/CD155 density favors TIGIT binding over DNAM-1 due to affinity differences. TIGIT blockade can shift that balance — but only if DNAM-1 is present to receive the signal. Without DNAM-1 competence, ligand density becomes an empty variable. The gate may be opened, but there is no effector machinery on the other side. This is why ligand-only biomarker strategies failed to stratify responders: they measured the brake, not the engine.
DNAM-1 Competence Is the Missing Biomarker
One of the quiet but fatal errors in TIGIT’s clinical development was not antibody design, dose, or even combination strategy — it was biomarker misassignment. TIGIT programs inherited PD-1 logic by default, enriching patients based on PD-L1 expression or prior checkpoint sensitivity, implicitly assuming that the same markers of exhaustion would predict response. The mechanistic literature now makes clear that this assumption was flawed from the outset. TIGIT does not operate on exhausted cells in isolation; it operates on DNAM-1–competent effectors whose function is being actively suppressed at the nectin gate.
Across preclinical and translational studies, a consistent signal emerges: CD226 (DNAM-1) expression is required for TIGIT blockade to restore cytotoxic function. TIGIT engagement suppresses DNAM-1 signaling through shared ligands (PVR/CD155 and CD112), receptor competition, and downstream inhibitory cascades. Blocking TIGIT removes this suppression — but only if DNAM-1 signaling machinery is intact. In systems where DNAM-1 is downregulated, uncoupled, or absent, TIGIT blockade has nothing to release. The brake is removed, but the engine is gone.
This explains why PD-L1 was the wrong enrichment marker. PD-L1 reflects interferon exposure and checkpoint engagement, not effector competence. A tumor can be PD-L1–high and yet populated by exhausted, terminally differentiated, or DNAM-1–deficient CD8 and NK cells incapable of responding even when TIGIT is blocked. Conversely, DNAM-1–competent effectors can exist in PD-L1–low settings, where TIGIT blockade yields minimal benefit because the inhibitory ligand density is insufficient to matter. Without DNAM-1 context, ligand expression alone is biologically ambiguous.
Critically, ligand density (PVR/CD155 or CD112) only becomes meaningful once DNAM-1 competence is established. High ligand density in a DNAM-1–low system represents a closed gate with no executable potential. High ligand density in a DNAM-1–high system represents precisely the condition where TIGIT blockade should unlock function. This reframes TIGIT responsiveness as a two-variable system — effector substrate availability on one axis, ligand geography on the other — rather than a single-marker exhaustion problem.
The failure to recognize DNAM-1 competence as the primary biomarker led to late-line TIGIT trials being populated with patients who violated the therapy’s core assumptions. Prior checkpoint exposure, chronic antigen stimulation, lymph node collapse, and progenitor exhaustion progressively erode DNAM-1–positive effector pools. By the time TIGIT was deployed, the substrate it requires had already disappeared. The biology did not fail — the patient selection did.
This insight also resolves why mechanistic consistency existed even as clinical results diverged. TIGIT blockade behaved exactly as expected in systems where DNAM-1 signaling was preserved and irrelevant where it was not. Once DNAM-1 competence is recognized as the gating variable, TIGIT’s mixed trial history collapses into a coherent pattern rather than a paradox.
Most importantly, this reframing opens a path forward. DNAM-1 competence is not merely a static biomarker — it is a repairable state. Therapies that expand or replenish DNAM-1–positive effector populations upstream transform TIGIT from an inert checkpoint into a functional amplifier. In that context, TIGIT does not create immune activity; it releases activity that already exists but is restrained. That distinction is foundational — and it is the bridge between TIGIT’s past failures and its potential future relevance.
As TIGIT trials began to fail, one explanation quickly rose to the surface: the antibodies themselves were harming the system. Early-generation TIGIT antibodies retained Fc activity, raising concerns about antibody-dependent cellular cytotoxicity (ADCC) or phagocytosis against TIGIT-expressing T and NK cells — precisely the populations the therapy was meant to support. In response, the field pivoted toward Fc-silent or Fc-null designs, removing this obvious self-inflicted wound. Mechanistically, this correction was real, justified, and necessary. But it was also fundamentally incomplete.
Fc-silencing solved a negative problem — it stopped TIGIT antibodies from actively depleting effectors — but it did nothing to address a positive requirement: the presence of DNAM-1–competent effector substrate in the first place. In other words, Fc-silent antibodies stopped making things worse; they did not make TIGIT work. Fc-engineering removed a harm vector but left intact the primary failure: absence of renewable DNAM-1-competent effectors
This distinction matters because it explains why Fc-optimized programs still produced disappointing results. Removing ADCC preserved whatever effector cells remained, but preservation is not restoration. In late-line patients — particularly those previously exposed to PD-1 blockade — effector pools are not merely fragile; they are often collapsed. TCF1⁺ progenitors are depleted, DNAM-1 expression is downregulated, and trafficking from tumor-draining lymph nodes is impaired. Under those conditions, a perfectly engineered TIGIT antibody is still acting on an empty system.
The newer mechanistic data sharpen this point. TIGIT blockade does not independently generate cytotoxicity; it releases DNAM-1 signaling from suppression at the nectin gate. If DNAM-1 signaling is absent or incompetent, there is nothing to release. Fc-silencing ensures that TIGIT blockade does not destroy effectors — but it cannot conjure them into existence, nor can it reconstitute progenitor-to-effector flow.
This explains a subtle but important pattern across TIGIT development: engineering refinements produced cleaner biology without producing durable efficacy. Trials became safer, signals became less noisy, but response rates did not meaningfully improve. The problem was no longer antibody harm — it was substrate absence.
Seen through this lens, Fc-silent design should be understood as a baseline requirement, not a solution. It removes a confounder so that TIGIT’s true dependency structure becomes visible. And once that structure is visible, the conclusion is unavoidable: TIGIT is an executional amplifier, not a regenerative therapy. Without upstream repair of DNAM-1–competent effector pools, even the best-designed TIGIT antibody is mechanistically stranded.
This is precisely where the TIGIT field stalled — and why further antibody tweaks alone were unlikely to succeed. The remaining bottleneck was no longer molecular; it was systemic.
DNAM-1 Competence: The Biomarker TIGIT Trials Never Measured
One of the quiet failures of the TIGIT development arc was not statistical, regulatory, or even clinical. It was diagnostic. TIGIT trials selected the wrong biomarker. DNAM-1 competence is the missing operational biomarker that bridges stemness and execution
From the outset, most TIGIT programs inherited enrichment logic directly from PD-1: PD-L1 expression, inflamed gene signatures, exhausted T-cell phenotypes, or prior checkpoint responsiveness. These markers made intuitive sense within a PD-1 worldview, where reinvigorating dysfunctional effectors is often sufficient to generate activity.
But TIGIT does not operate on the same axis. TIGIT does not primarily modulate exhaustion. It modulates competitive signaling between inhibitory and activating receptors that share the same ligands — most importantly, TIGIT versus DNAM-1 (CD226) at CD155 (PVR) and CD112. That distinction changes everything.
Beyond TCF1⁺ Progenitors: DNAM-1⁺ Effector Expansion Key Part Of Substrate Creation
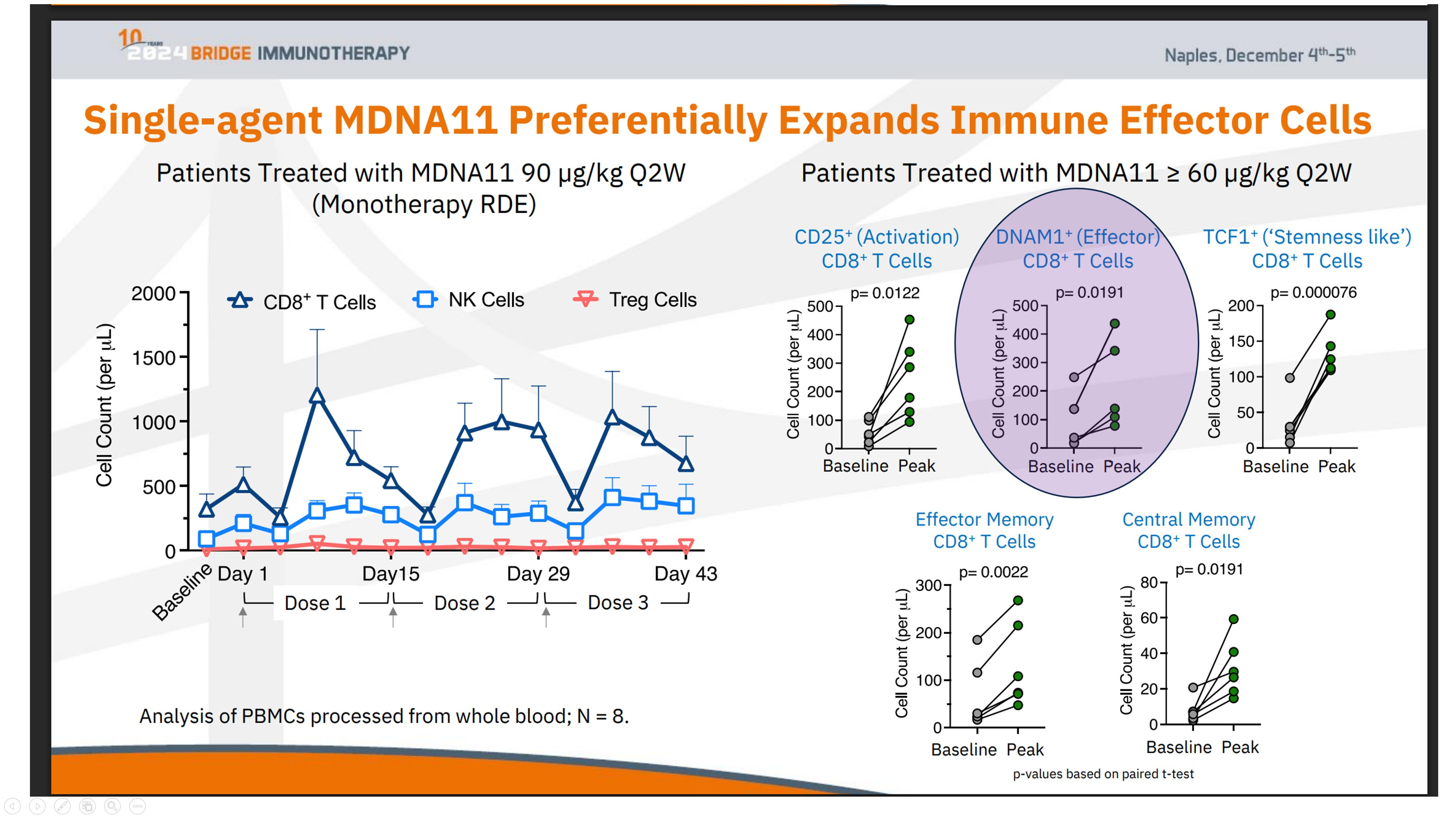
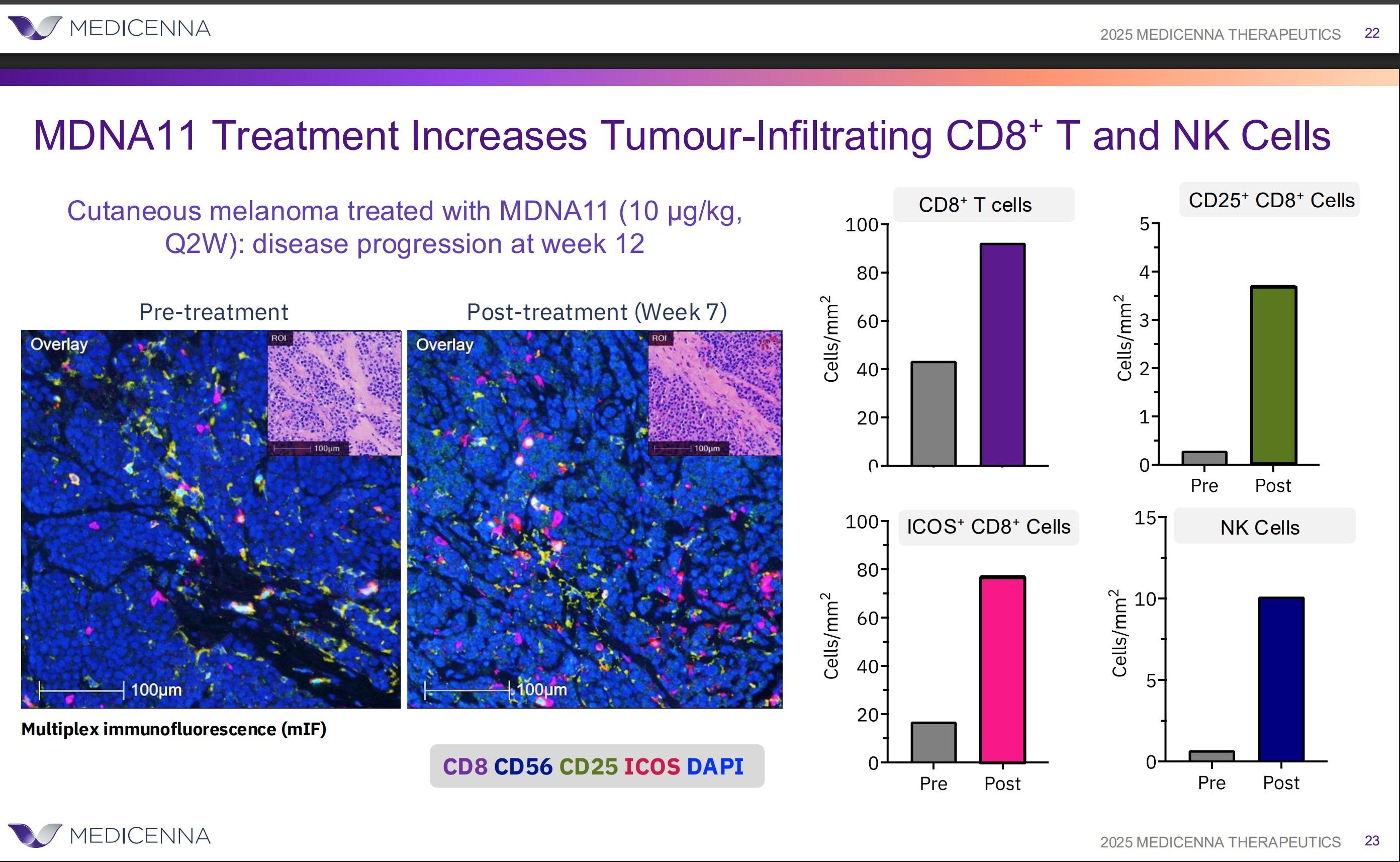
The expansion of DNAM1⁺ (CD226⁺) CD8⁺ T cells, as observed in relevant data (see the chart above), provides a key insight into this repair process. DNAM-1 is more than a simple activation marker; it serves as the functional counterweight to inhibitory axes like TIGIT. It is selectively lost when immune systems are pushed into terminal exhaustion, often as a downstream effect of failed priming.
What sets the DNAM1⁺ expansion in the ABILITY-1 context apart is not just the growth of these effector CD8⁺ T cells, but their parallel preservation alongside upstream progenitor compartments—rather than at the progenitors’ expense. In most late-line immunotherapy settings, DNAM-1 biology falters because the lymph-node renewal loop has already broken down. Attempting to block TIGIT in such environments cannot restore function, as the essential substrate is absent.
This observation reframes the clinical failure of TIGIT programs not as a dismissal of the TIGIT/CD226 axis, but as evidence of its dependency on upstream integrity. TIGIT blockade relies on renewable CD226⁺ effector populations, which MDNA11 helps restore. Thus, the DNAM1⁺ expansion signals not downstream amplification, but successful repair of upstream immune assembly, enabling functional effectorization. In essence, this data does not position MDNA11 as a “better” alternative to TIGIT; it clarifies why TIGIT lacked viability in populations where lymph-node priming had collapsed, highlighting therapies that operate upstream as a distinct category.
Preservation of CD8⁺ stem-like and NK compartments following upstream repair.
These data illustrate that immune expansion following MDNA11 is not consumptive. Progenitor-linked and DNAM-1⁺ effector populations are preserved in parallel, maintaining renewable immune capacity rather than depleting it.
The Missing Diagnostic Frame
Put plainly, TIGIT trials asked the wrong question. They asked: Is the tumor expressing the right ligands? They should have asked: Is the immune system still capable of using them?
DNAM-1 competence — across CD8⁺ effector, memory, and NK compartments — answers that question directly. It integrates immune history, lineage integrity, and functional readiness into a single, biologically coherent marker. This also reframes future trial design. TIGIT does not belong in indiscriminate post-PD-1 salvage. It belongs in systems where DNAM-1⁺ effector supply exists or can be restored.
Which brings the argument full circle — and sets up the central insight of the article: TIGIT was never a standalone therapy. It was always a downstream amplifier, waiting for upstream repair.
The Hidden Signal in the Failures
In hindsight, the persistence of TIGIT failure even after Fc correction is not discouraging — it is diagnostic. It tells us that:
TIGIT’s biology was not wrong
The checkpoint was not redundant
The ligand axis remained intact
What was missing was substrate. Once that insight is accepted, the data stop looking contradictory. They look inevitable. TIGIT blockade did not fail because antibodies were poorly designed. It failed because it was deployed after immune assembly had already collapsed.
And that realization sets up the critical transition to the NES framework — where TIGIT is no longer evaluated as a standalone therapy, but as a conditional amplifier that only works once immune structure has been rebuilt.
Post-Mortem of TIGIT/CD226
What Failed — and What That Failure Actually Proved
The widespread clinical failure of TIGIT-targeting antibodies is often treated as a cautionary tale about over-interpreting checkpoint biology. Read through a lymph-node priming lens, however, TIGIT’s collapse tells a far more precise story: checkpoint modulation failed not because the TIGIT/CD226 axis was irrelevant, but because it was applied downstream of a more fundamental immune assembly failure.
TIGIT blockade was explicitly designed to “release” functional CD226 (DNAM-1)–expressing CD8⁺ effector T cells from inhibitory signaling. That premise holds only if a renewable CD226⁺ effector substrate is continuously being generated upstream. In CPI-refractory colorectal cancer populations, that assumption repeatedly proved false.
Despite clean pharmacology, acceptable safety profiles, and clear on-target engagement, multiple late-stage TIGIT programs failed to produce durable or reproducible benefit. This discrepancy resists explanation through tumor-centric or checkpoint-centric frameworks alone. It aligns cleanly, however, with a model in which tumor-draining lymph-node (TDLN) architecture and TCF1⁺ progenitor renewal have already collapsed.
Just as Node Extension Scores reframed PD-1 efficacy as a function of immune reach, the TIGIT experience suggests the need for an analogous construct — a way to quantify whether the DNAM-1⁺ substrate required for TIGIT resuscitation even exists. Absent that substrate, failure is not surprising; given the context PRISM-11 has translationally explained it is inevitable.
Loss of CD226 expression is not the initiating defect — it is the downstream consequence of failed progenitor maintenance and disrupted lymph-node immune assembly. When TCF1⁺ progenitors are no longer renewing, effector populations cease to regenerate. In that context, TIGIT blockade cannot “restore” function, because the biological substrate it depends on is no longer being replenished.
Seen this way, TIGIT’s clinical failure does not invalidate the TIGIT/CD226 axis. It validates a more fundamental ordering principle: durable immunotherapy requires restoration of lymph-node–resident immune renewal before modulation of terminal effector signaling can succeed. Checkpoint precision cannot substitute for immune assembly which is what MDNA11 is showing you as evidenced by this cut out from their data.
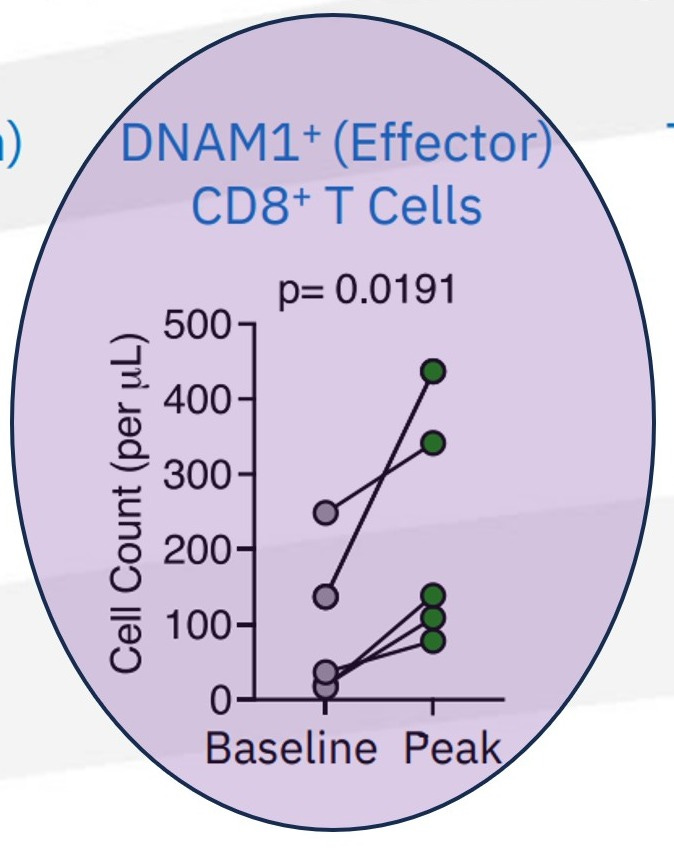
Just as Node Extension Scores reframed PD-1 efficacy as a function of immune reach, the TIGIT experience suggests the need for an analogous construct — a way to quantify whether the DNAM-1⁺ substrate required for TIGIT resuscitation even exists. Absent that substrate, failure is not surprising; given the context PRISM-11 has translationally explained it is inevitable.
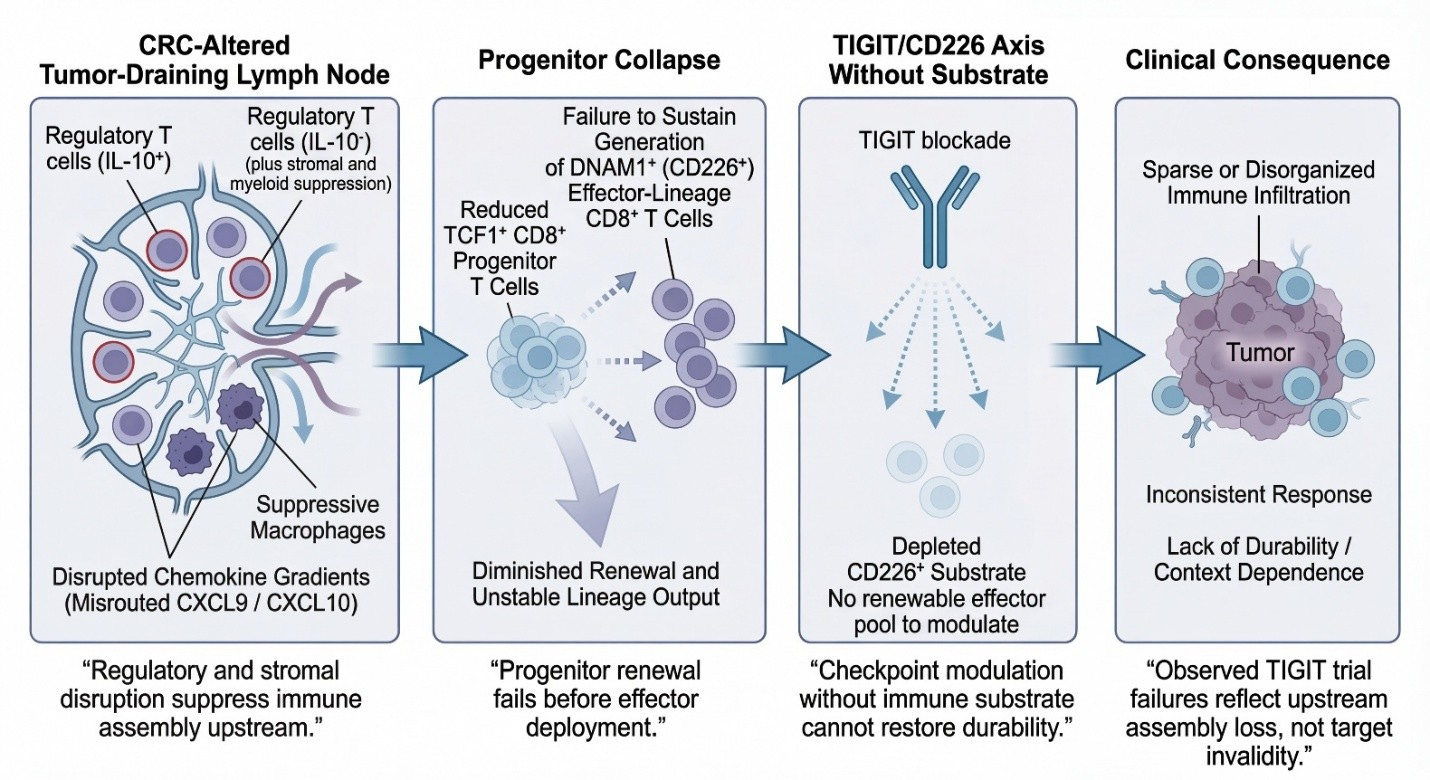
While the schematic below is drawn from colorectal cancer—where TDLN disruption, progenitor collapse, and checkpoint refractoriness are well characterized—the logic it depicts is not CRC-specific. It reflects a general failure mode of downstream checkpoint blockade when immune assembly has already collapsed. CRC is shown here because it makes the sequence visible, not because the mechanism is unique.
TIGIT as a Conditional Amplifier in the NES Framework
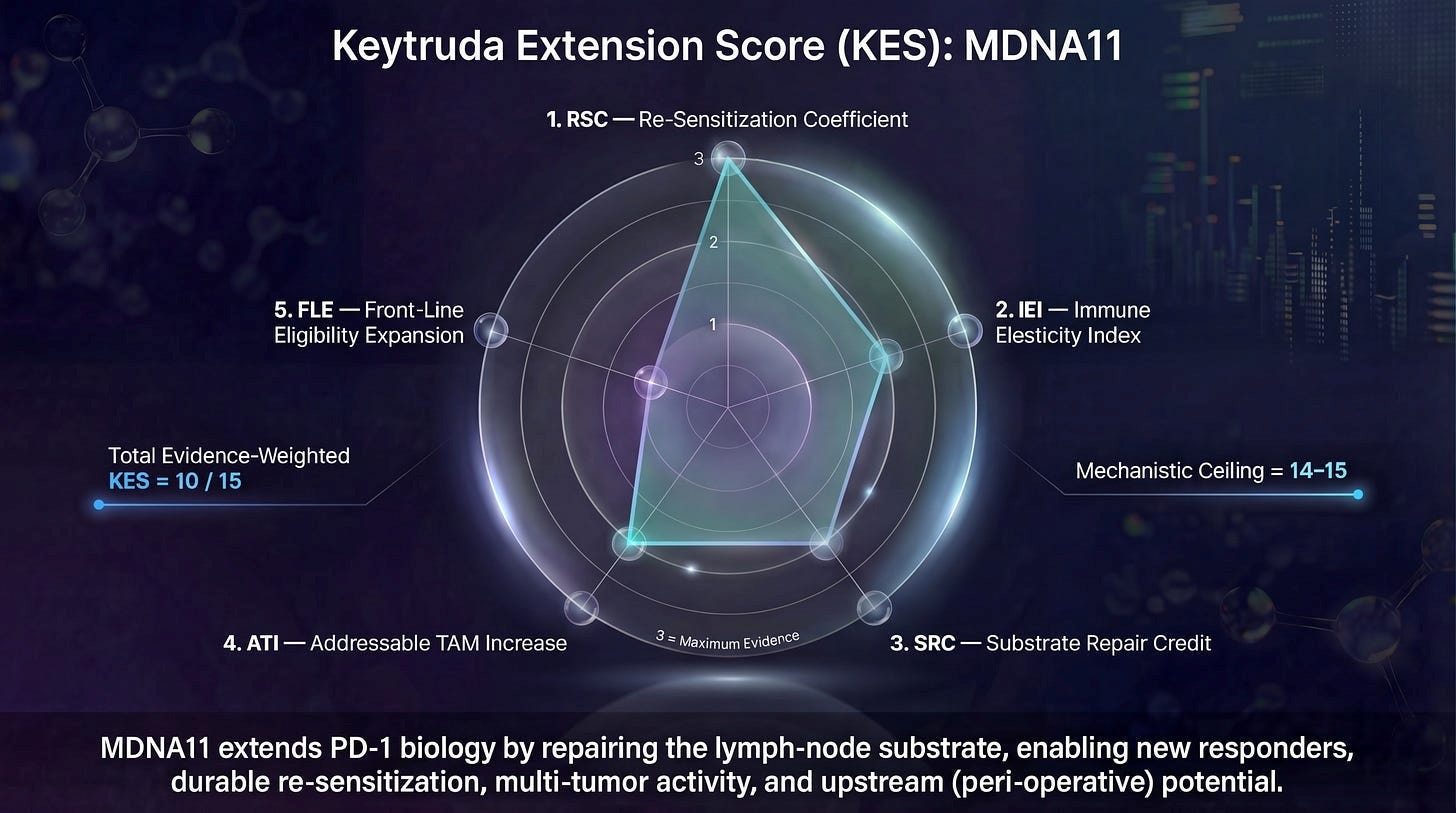
Now might be a good time to go back and read the NES substack article here. If you recall, this started out as the Keytruda Extension Score.
With this broad framing in mind, once TIGIT’s failures are reclassified and its biomarker dependency clarified, its proper place in the immune control architecture becomes unmistakable. TIGIT is not a foundational therapy. It is not a repair mechanism. It is not a substitute for immune assembly. TIGIT is a conditional executional amplifier — a therapy whose relevance emerges only after upstream immune continuity has been restored.
This is precisely the type of mechanism the Node Extension Score (NES) was designed to distinguish. Within the NES framework, therapies are not judged by whether tumors shrink in their presence, but by which layer of immune continuity they causally repair or enable. That distinction is critical, because multiple interventions can produce similar downstream readouts while contributing entirely different types of value — or fragility — to the system.
TIGIT earns no Substrate Repair Credit (SRC). It does not rebuild TCF1⁺ progenitor pools, restore lymph node architecture, or convert consumptive immune responses into renewable ones. It also earns no Immune Extension & Integrity Credit (IEI). TIGIT does not repair trafficking, address vascular exclusion, or ensure that effector cells can reliably reach and persist at the tumor site. These functions must already be in place before TIGIT can act.
What TIGIT does earn — under the right conditions — is Re-sensitization Credit (RSC). By interrupting inhibitory signaling at the nectin gate, TIGIT blockade can restore DNAM-1–mediated cytotoxic function if and only if DNAM-1–competent effector cells are present and properly deployed. In NES terms, TIGIT unlocks executional potential that already exists; it does not create it.
This distinction explains why TIGIT’s clinical history appears so inconsistent when viewed through traditional response-based lenses, yet becomes coherent when mapped onto immune continuity. In early-line or less-damaged systems, where substrate and extension remain partially intact, TIGIT occasionally produced incremental benefit. In late-line, substrate-depleted systems, TIGIT failed catastrophically — not because its target was invalid, but because the immune system no longer possessed the capacity to express executional gains.
The NES framework therefore reframes TIGIT’s trajectory as a sequencing error, not a scientific one. TIGIT was repeatedly deployed in contexts where RSC could not be expressed because SRC and IEI had already collapsed. No amount of antibody optimization could overcome that structural limitation.
This is also where MDNA11 enters the picture with clarity rather than hype. MDNA11 operates upstream of TIGIT’s failure mode. By engaging tumor-draining lymph nodes and preferentially expanding DNAM-1⁺ effector and memory-biased CD8 populations, MDNA11 supplies SRC and supports the conditions necessary for IEI. In doing so, it restores the immune substrate TIGIT depends on to function. TIGIT does not “synergize” with MDNA11 in the colloquial sense; it becomes eligible to work only because MDNA11 has repaired the system.
In NES terms, this is not additive biology — it is hierarchical enablement. MDNA11 rebuilds immune continuity. TIGIT then amplifies execution within that restored architecture. Confusing these roles leads to wasted capital and failed trials; respecting them allows downstream amplifiers to be deployed where they actually matter.
Seen this way, TIGIT is not a failed checkpoint. It is the canonical example of why executional therapies must be sequenced after repair, not instead of it. And it demonstrates exactly why the NES Expansion Map exists: to prevent biologically valid mechanisms from being discarded simply because they were applied to a system that could no longer respond.
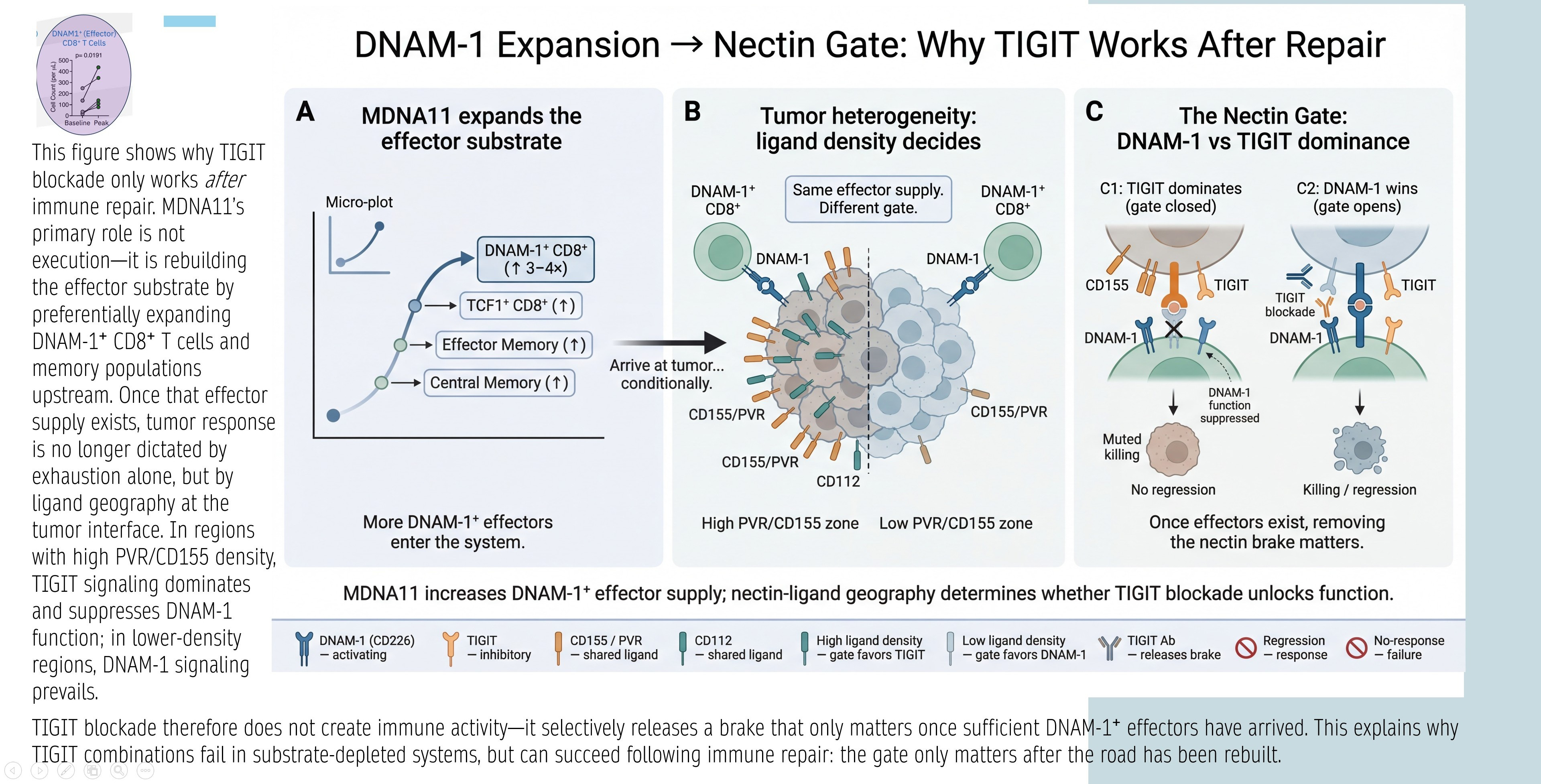
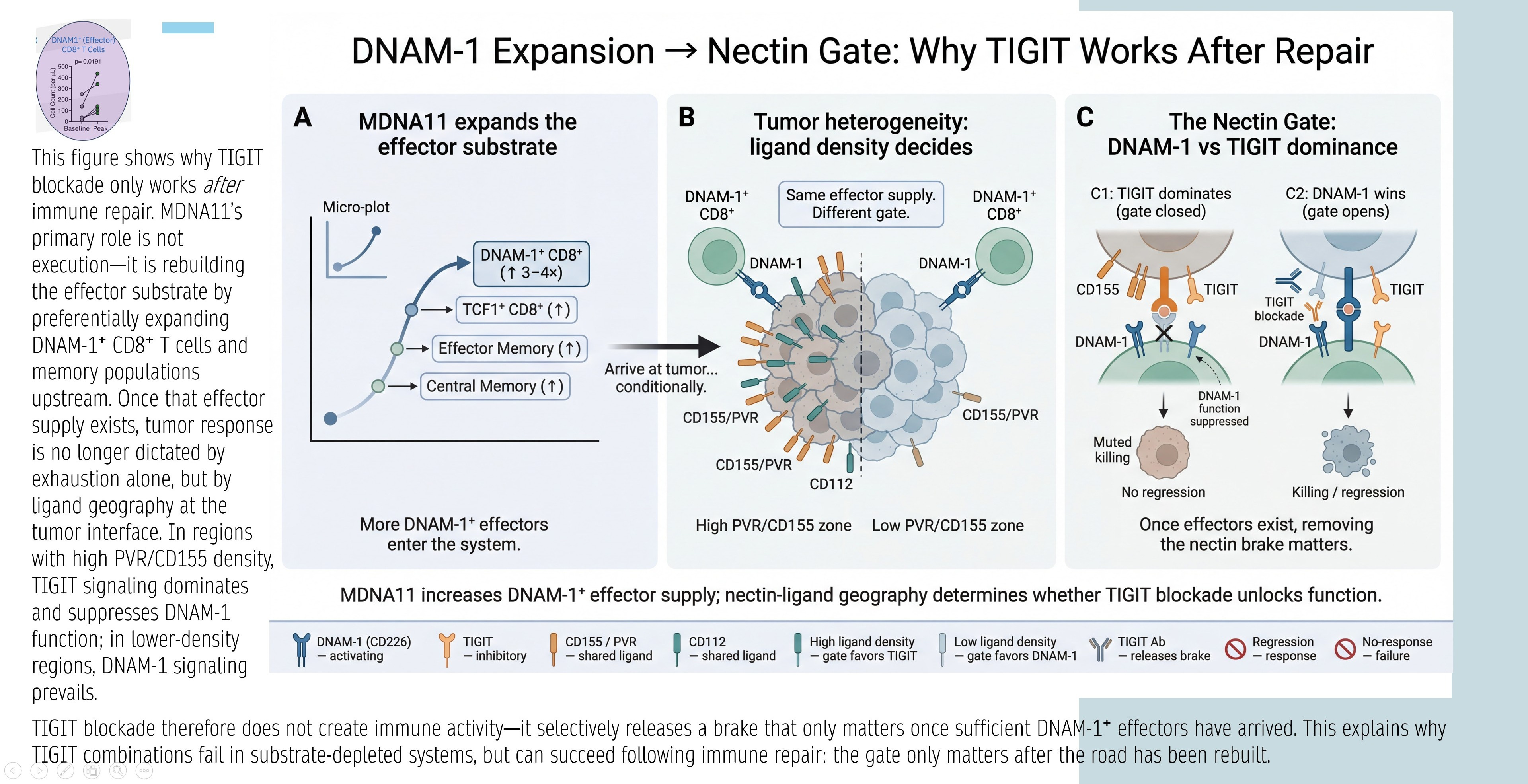
DNAM-1 Expansion → Nectin Gate: Why TIGIT Works After Repair
The graphic below explains TIGIT’s conditional efficacy: TIGIT blockade releases a “nectin brake” only if sufficient DNAM-1+ effectors exist. Panels:
A: MDNA11 expands DNAM-1+ CD8⁺ substrate (TCF1⁺ progenitors → effectors/central memory).
B: Tumor heterogeneity/ligand density decides gate: High PVR/CD155 favors TIGIT inhibition; low favors DNAM-1 activation.
C: Nectin Gate dynamics— TIGIT dominates (closed gate, no response) unless DNAM-1 supply overwhelms (open gate, killing/regression).
Synthesis: TIGIT fails in depleted systems; succeeds after repair (MDNA11 increases DNAM-1+ effectors, shifting geography favor).
Relevance: Directly ties MDNA11 to TIGIT rejuvenation—Layer 2 repair supplies DNAM-1+ for Layer 4 nectin interactions.
The structural upgrade: re-map your synthesis into NES terms
If you rewrite the synthesis in NES-native language, you get a clean bridge from “TIGIT gate” → “NES Expansion Map.” Here’s the framing:
SRC (Layer 2): MDNA11 expands/renews a DNAM-1⁺ effector-capable reservoir (plus memory scaffolding).
IEI (Layer 3): if trafficking/addressins/chemokines/vasculature are broken, the DNAM-1 reservoir never expresses at the tumor — TIGIT is irrelevant.
RSC (Layer 4): once DNAM-1⁺ effectors are present at tumor, nectin ligand density becomes a true gate; TIGIT blockade is now meaningful.
ATI/FLE: durability and line-of-therapy portability determine whether the “TIGIT rescue” is a one-off or a scalable platform move.
Let’s do a little side bar here as there’s a lot going on! First, lets define:

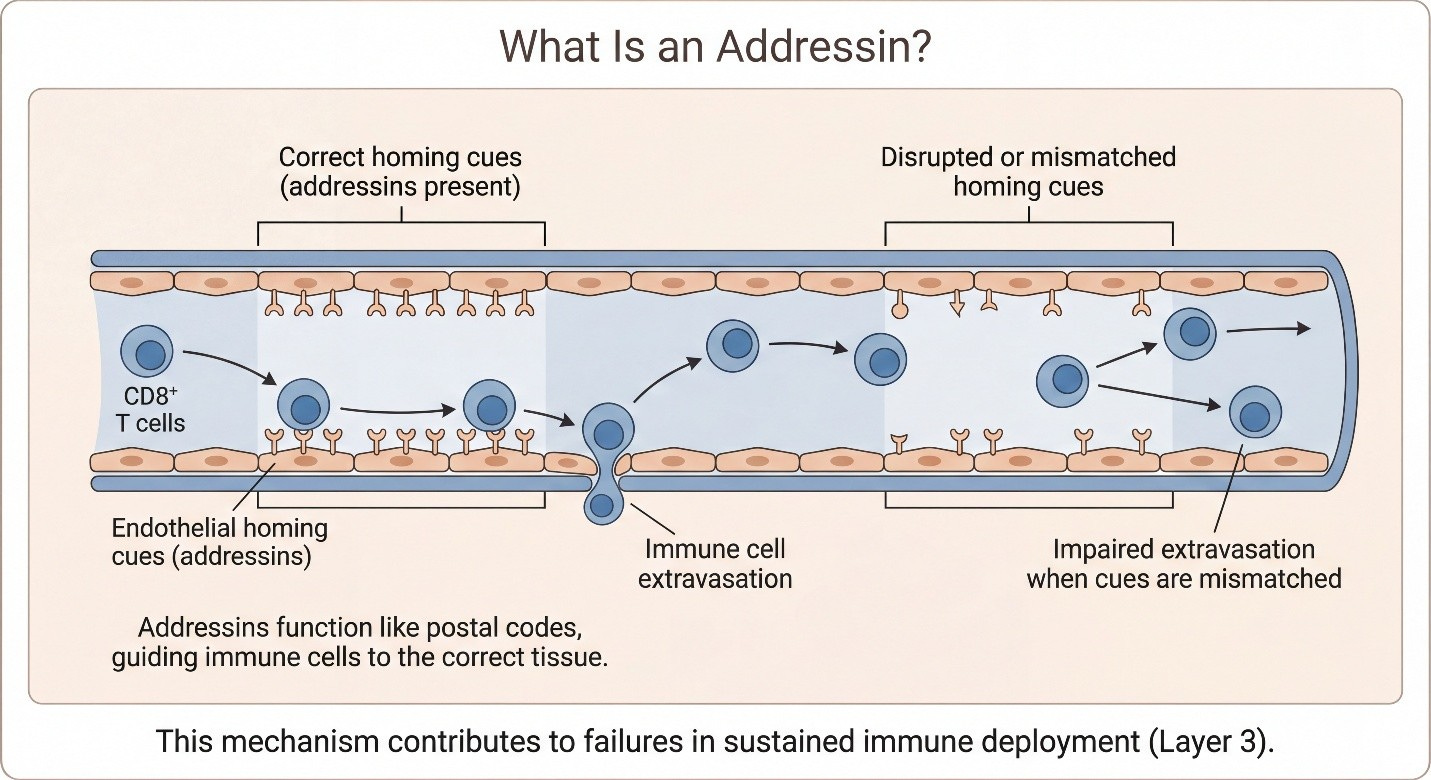
Introducing MDNA11 here is not a product detour. It serves a narrower purpose: to show that TIGIT’s conditional requirements are not theoretical—they can be re-established when upstream immune assembly is repaired. The point is not that TIGIT needs “a better TIGIT.” The point is that TIGIT needs a system where amplification is once again biologically meaningful.
MDNA11 as an Enabler (Not the Hero)
What MDNA11 Can Do For TIGIT
The TIGIT (T cell immunoreceptor with Ig and ITIM domains) target, an immune checkpoint protein, was first identified by Genentech (Roche) in 2008 and published in 2009. It emerged as a promising immunotherapy focus around 2018–2020, driven by early clinical data suggesting it could enhance PD-1/PD-L1 inhibitors in cancers like non-small cell lung cancer (NSCLC) and gastrointestinal tumors. Investments ramped up significantly from 2020 onward, as TIGIT was hailed as a potential “next PD-1” in immuno-oncology. However, the field has seen multiple clinical setbacks since 2022, leading to program terminations by companies like Bristol Myers Squibb (BMS), GSK, and Novartis, with many milestones unpaid and investments unlikely to be recouped. Over the past 15 years (roughly 2011–2026), TIGIT investments in biotech and pharma have totaled several billion dollars, concentrated in the last 6–8 years. This includes upfront licensing payments, equity investments, milestones (largely contingent and unpaid), and R&D costs. Below is a breakdown based on major deals and programs, drawing from industry reports and company disclosures. Note that exact totals are challenging to pinpoint due to opaque R&D budgets, but estimates suggest $5–10 billion+ across upfronts, equity, milestones, and development (with many programs failing to advance).
Where MDNA11 Fits—and Why This Position Is Structurally Different
In contrast to these downstream-focused therapies, MDNA11 occupies a unique upstream position by design. As a β-selective IL-2 agonist delivered systemically and rhythmically, it engages and generates lymph node-resident TCF1⁺ progenitor populations, reinforcing STAT5-mediated renewal. This approach prioritizes repairing immune capacity before deployment, rather than solely expanding terminal effectors. As a result, it enhances subsequent checkpoint sensitivity and supports sustained waves of effector cells. MDNA11 is not merely an amplifier of downstream immunity; it functions as a repair mechanism for immune assembly itself. This distinction is crucial, as it directly influences the potential for achieving durability in immunotherapy outcomes.
MDNA11’s Unique Fit: Reviving TIGIT via Upstream Repair
The PRISM-11 slide brilliantly illustrates why MDNA11 could “rescue” TIGIT: It expands DNAM-1+ effector substrate (Panel A: TCF1⁺ progenitors → central/effector memory via β-IL-2), shifting tumor nectin geography (Panel B: ligand density decides gate) to favor DNAM-1 dominance over TIGIT (Panel C: open gate enables killing/regression).
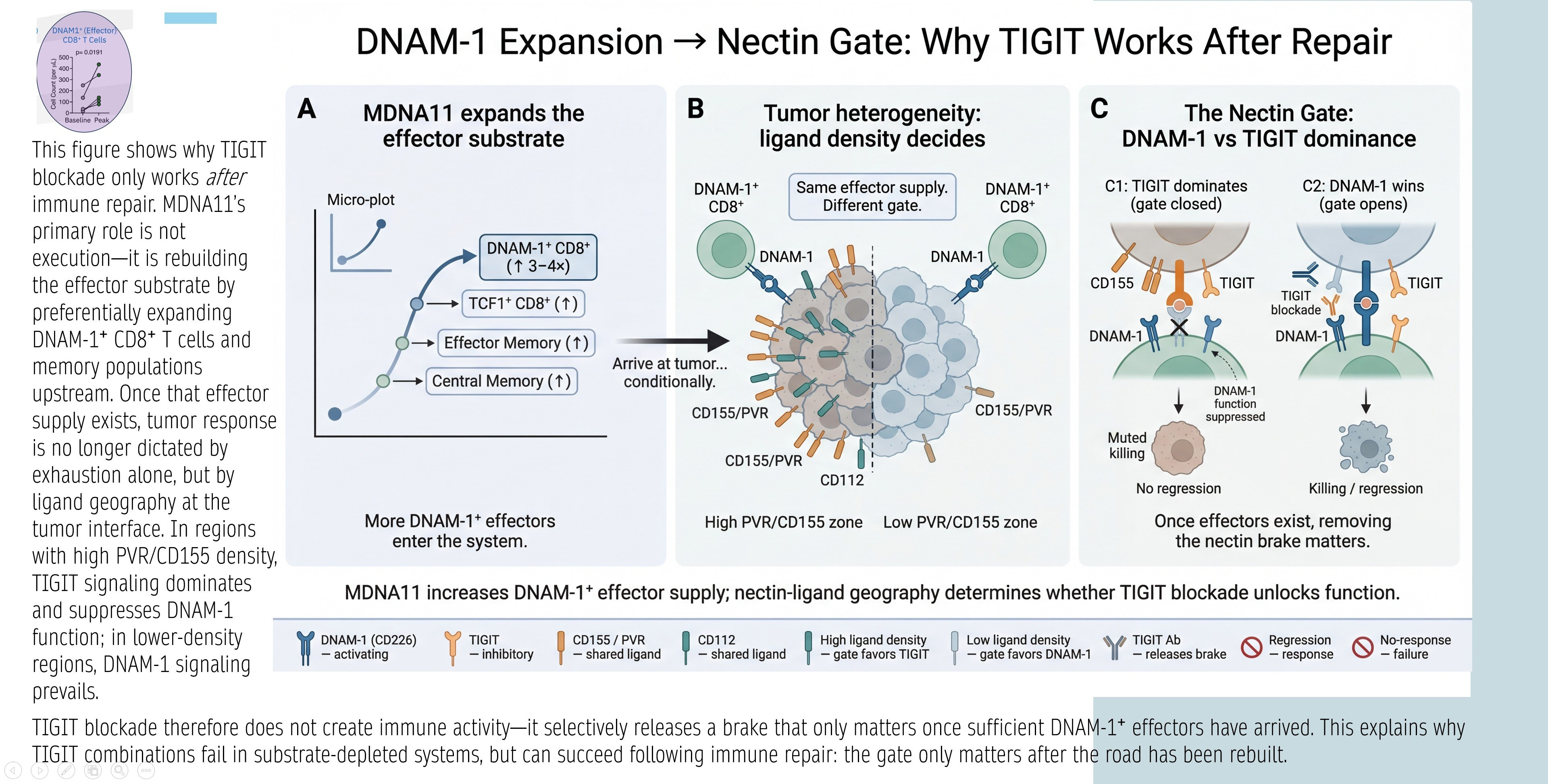
TIGIT fails in depleted systems but succeeds “after repair”—MDNA11 provides the Layer 2 assembly (rhythmic STAT5 on LN-resident progenitors) to flip the nectin brake. This isn’t speculative: ABILITY-1 data (ESMO-IO 2025) shows TCF1⁺ persistence as LN priming signal, with durability in post-ICI failures—precisely where TIGIT combos struggle (e.g., Roche’s tiragolumab + Tecentriq showed only 20-30% ORR in resistant NSCLC). By replenishing DNAM-1+ pools, MDNA11 could enable TIGIT in high-PVR tumors (nectins paper: PVR/CD155 on tumor/CAFs suppresses via TIGIT/CD96), turning transient responses into durable control.
Synthesis: MDNA11’s Role in Rejuvenating TIGIT Space
Across independent lines of investigation, the TIGIT–nectin axis emerges as a central immune evasion circuit. Nectins and related ligands (CD155/PVR, CD112) bind TIGIT, CD96, and PVRIG, tilting the balance away from DNAM-1 (CD226)–mediated activation. In physiologic settings, this axis regulates effector restraint. In tumors with high PVR/CD155 dominance and depleted effector pools, it becomes a functional brake layered onto an already compromised immune substrate.
The clinical failures of TIGIT programs are therefore less about target invalidity and more about substrate absence. Multiple late-line studies deployed TIGIT blockade into settings characterized by prior PD-1 exposure, exhausted CD8⁺ pools, and disrupted tumor-draining lymph node (TDLN) architecture. In such environments, DNAM-1⁺ effector competence is frequently diminished. Blocking TIGIT under those conditions removes an inhibitory signal from a pathway that is already non-functional.
MDNA11 alters this equation upstream. As a βγ-selective IL-2 superkine with Fc-albumin–mediated lymph node residency, MDNA11 drives rhythmic STAT5 activation within the TDLN, expanding TCF1⁺ progenitor CD8⁺ cells and restoring DNAM-1⁺ effector substrate. Rather than acting at the point of exhaustion, it rebuilds the renewable immune supply chain. Once DNAM-1 dominance is re-established, TIGIT blockade regains functional relevance in high-ligand tumors.
In Control Stack terms, TIGIT operates at Layer 4—effector inhibition within infiltrated T and NK compartments. The nectin axis exerts governance pressure across the tumor microenvironment, influencing stromal, endothelial, and immune interfaces (Layer 1–1.5). MDNA11 functions at Layer 2, supplying DNAM-1⁺ substrate capable of contesting nectin-mediated suppression. Under this architecture, TIGIT does not create capacity; it amplifies capacity that has been structurally restored.
This reframing suggests that TIGIT’s commercial retrenchment reflects mis-sequencing rather than terminal target failure. If deployed after upstream immune repair—rather than into late-line immune collapse—the axis may yet demonstrate meaningful activity in ligand-high NSCLC and gastrointestinal tumors. Emerging bispecific approaches targeting both nectin ligands and TIGIT further reinforce the idea that the gate is biologically active, but contingent on effector competence.
Editor’s Note (February 26, 2026):
The synthesis section was lightly edited to remove internal drafting references (e.g., “per DOCX,” “per paper”) and replace them with direct narrative framing aligned to the published references section. No substantive analytical changes were made.
References
TIGIT / CD226 (DNAM-1) / PVR (CD155) axis — mechanism, gate logic, and why CD226 competence is prerequisite
Jin HS, Ko M, Choi DS, et al. CD226hi CD8+ T Cells Are a Prerequisite for Anti-TIGIT Immunotherapy. Cancer Immunology Research. 2020.
Johnston RJ, Comps-Agrar L, Hackney J, et al. The TIGIT/CD226 axis regulates human T cell function. The Journal of Immunology. 2014.
Ge Z, Peppelenbosch MP, Sprengers D, Kwekkeboom J. TIGIT/CD226 Axis Regulates Anti-Tumor Immunity. Pharmaceuticals (Basel). 2021.
Harjunpää H, Guillerey C. Emergence of the CD226 Axis in Cancer Immunotherapy. Frontiers in Immunology. 2022.
Chauvin JM, Zarour HM. TIGIT-CD226-PVR axis: advancing immune checkpoint blockade for cancer immunotherapy. Journal for ImmunoTherapy of Cancer. 2022.
Deng W, Gowen BG, Zhang L, et al. CD226: a potent driver of antitumor immunity that needs to be protected from immune checkpoint inhibition. Cellular & Molecular Immunology. 2021.
Maruhashi T, Sugiura D, Okazaki IM, Okazaki T. Mechanistic convergence of the TIGIT and PD-1 inhibitory pathways on CD226. Immunity. 2022.
TCF1⁺ progenitor pools, exhaustion architecture, and why downstream checkpoint response depends on renewable substrate
Chen Z, Ji Z, Ngiow SF, et al. TCF-1-Centered Transcriptional Network Drives an Effector versus Exhausted CD8 T Cell Fate Decision. Immunity. 2019.
Sade-Feldman M, Yizhak K, Bjorgaard SL, et al. TCF1 maintains CD8+ T cell stemness in tumor microenvironment. Journal of Leukocyte Biology. 2021.
Guo X, Ma S, Wang J, et al. Terminally exhausted CD8+ T cells in solid tumors: biology, biomarker potential and translational tools. Frontiers in Immunology. 2025.
Brandl A, Daum R, Dillinger B, et al. TCF1+PD-1+ tumour-infiltrating lymphocytes predict response to immune checkpoint inhibition in NSCLC. Lung Cancer. 2022.
Luoma AM, Suo S, Wang Y, et al. Stem-like CD8+ T cells in cancer. Frontiers in Immunology. 2024.
Tumor-draining lymph node (TDLN) integrity as the upstream reservoir that feeds systemic and intratumoral response
Fransen MF, Schoonderwoerd MJA, Ossendorp F. The tumor-draining lymph node as a reservoir for systemic immune responses and immunotherapy efficacy. Trends in Cancer. 2023.
Disclosure Statement
The author is an investor in Medicenna Therapeutics and licenses the PRISM-11 analytical framework to Medicenna for internal and external use with attribution. The author is not an employee of Medicenna. All views expressed are independent and for informational purposes only.
David: Again, you so brilliantly lay this out! Here comes my “broken record “ again.
Why is it taking so long for the pharmaceutical industry to recognize it??? I understand there are more presentations in March, maybe that will help.
Also, with your new relationship with Medicenna, I would hope Fahar sends your stuff to the right people.
Ed
Fascinating series of articles. I've got some questions but I suspect they'll be addressed in the upcoming revised operational playbook article so I'll hold off until then. Oh, to be a fly on the wall in Medicenna's conference room.